



Introduction
Currently, the human body is viewed not only as a complex organism, but also as an environment within which countless microorganisms live. The number of microorganism genes in the human body is approximately 150 times the number of human genes (Ref. Reference Qin1). These microorganisms live and interact with the human body in different symbiotic relationships, for example, mutualism, commensalism or even parasitism (Ref. Reference Eloe-Fadrosh and Rasko2). These interactions may be associated with a disturbance in normal physiology. Therefore, microbiome studies may provide a better understanding of the development of human diseases.
The human stomach was once thought to be a ‘sterile’ environment because of its extremely high acidity (Refs Reference Nardone and Compare3, Reference Petra, Rus and Dumitraşcu4). It was then thought to be the most important gastrointestinal barrier to various pathogenic microorganisms. However, this theory was debunked when Helicobacter pylori was successfully isolated from the human stomach by Barry Marshal and Robin Warren in the early 1980s (Refs Reference Warren and Marshall5, Reference Marshall and Warren6). This breakthrough suggested that other microorganisms might be able to survive and colonise the human stomach. However, past efforts to profile stomach microbiota were hampered by the limitations of conventional culture, histology and immunohistochemistry. The culture method required a huge effort to isolate and identify bacteria and could not characterise most of them. Therefore, it most likely underestimated the actual number of microbiota in the stomach.
The development of sequencing technology has shed new light on the gastric microbiota. The application of next-generation sequencing (NGS) is a huge leap that has enabled a shift to a culture-independent approach and focused on sequence analysis to identify stomach microorganisms. The microbiome and transcriptome are then used to characterise the microbiota and viable microorganisms in the stomach.
Another type of study, the metabolome study, is currently used to support microbiome and transcriptome studies. A metabolome study is a comprehensive analysis of metabolites, where the molecules released by the organisms into the environment are identified and quantified. The metabolome is considered the most direct indicator of alterations in the environment.
In this review, we aim to summarise the current updates on microbiota, microbiome, transcriptome and metabolome studies of the human stomach. We also review the importance of integrating multiple datasets to get a better understanding of the development of gastric diseases.
Next-generation sequencing
The stomach is unique within the human gastrointestinal tract because of its harsh environment with extremely high acidity, ranging between a pH of 1 and 3 in healthy subjects (Refs Reference Ayazi7, Reference McLauchlan8). Therefore, it is extremely difficult for microorganisms to successfully colonise it. Most viable microorganisms are eliminated in the stomach, resulting in significantly lower microbial loads in the stomach (101–103 microbes/g) compared with that in the small intestine (104–107 microbes/g) or colon (1011–1012 microbes/g) (Refs Reference Berg9–Reference O'Hara and Shanahan11). This is one of the causes of the difficulty in profiling the microorganisms within the human stomach. In the past, very few methods could be used for gastric microbiota profiling. A conventional method such as the culture of gastric biopsy and gastric juice can reveal viable microorganisms in the specimens (Refs Reference Sanduleanu12–Reference Khosravi14). Histology and immunohistochemistry are also frequently used for profiling (Ref. Reference Sanduleanu12). However, these methods are costly, require extensive effort, take a long time to achieve results and have a low sensitivity. Therefore, a more reliable method to provide detailed data about gastric microbiota is needed. NGS has been used extensively for comprehensive profiling of microorganisms ever since the Human Microbiome Project began (Ref. Reference Peterson15). NGS is a ‘culture-independent’ method that can provide a deep and high throughput of DNA sequences. It results in gastric microbiome data, which then can be used to determine the microbiota. Microbiome, in this case, refers to the collection of genomes from all microorganisms in a specific environment, whereas microbiota refers to the specific microorganisms. The pipeline for a microbiome study is shown in Figure 1.
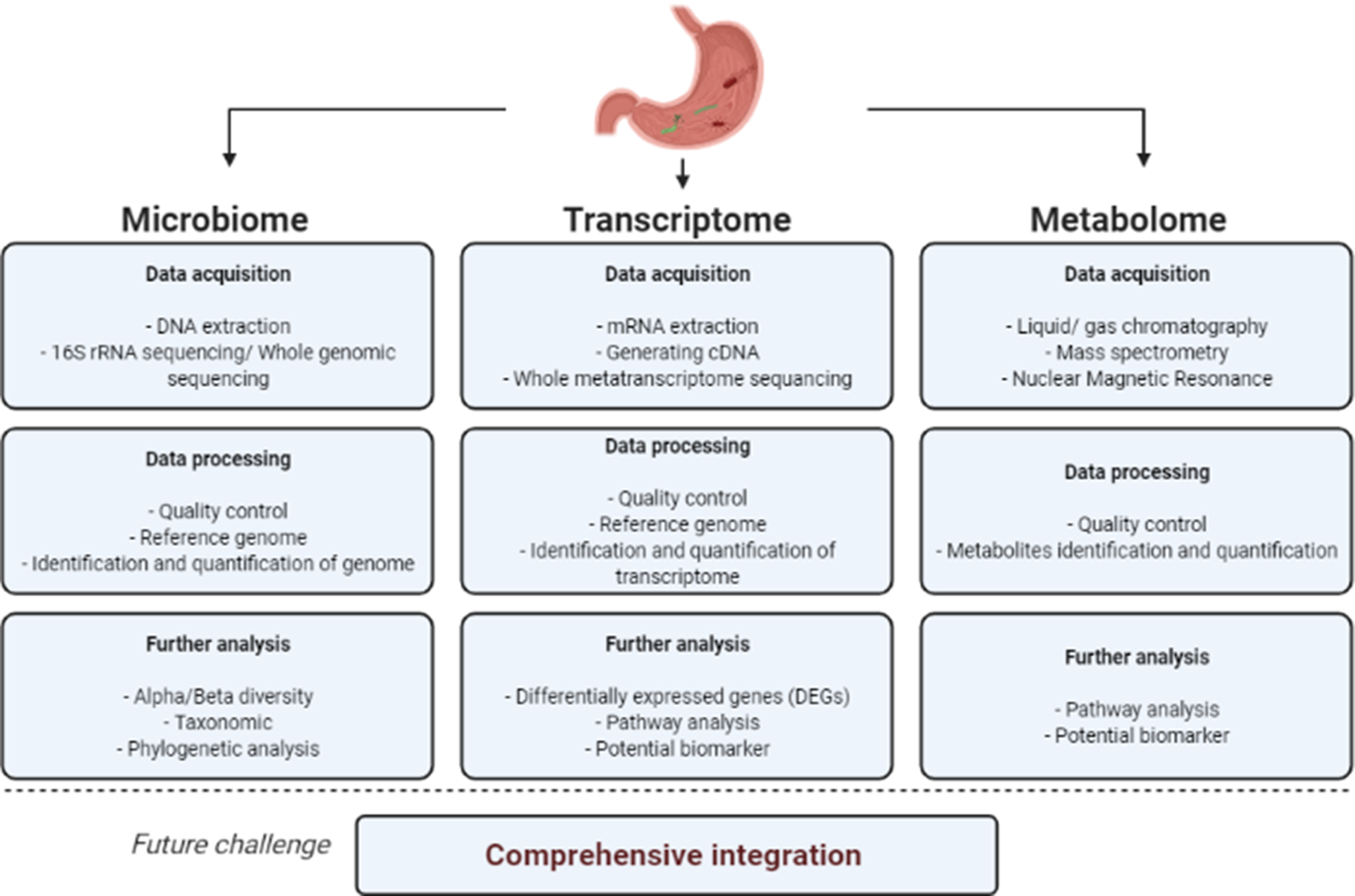
Fig. 1. Schematic pipeline for microbiome, transcriptome and metabolome study.
Currently, NGS is widely used for profiling microorganisms in a specific environment, such as the stomach, gut, oral cavity or skin. There are several NGS methods; however, 16S ribosomal RNA (16S rRNA) gene sequencing and shotgun metagenomic sequencing are the two most used. The 16S rRNA sequencing method uses 16S rRNA as a marker gene to be amplified. 16S rRNA is a highly conserved gene with hyper-variable regions within it; it is unique and can be used to distinguish between species and microbiome strains (Refs Reference Osman16, Reference Bharti and Grimm17). The hyper-variable regions within the 16S rRNA gene, such as V1–V9, are the amplification target in microbiome studies. A combination of several hyper-variable regions is also commonly used, such as the combination of V1–V3, V2–V3, V3–V4 and V5–V6 (Refs Reference Miftahussurur18–Reference Jo21). The sequencing reads are then filtered based on the number of expected errors and the length of the reads to obtain high-quality clean tags. Chimeric sequences can be detected using specific software such as UCHIME (Ref. Reference Edgar22). Chimeric sequences are usually removed to obtain the effective tag sequences and to avoid misidentification. Next, sequences with high similarity are assigned the same cluster name operational taxonomic unit (OTU). Most studies used a 97–99% similarity as the basis of determining the OTUs (Refs Reference Miftahussurur18, Reference Ferreira19, Reference Jackson23). However, 16S amplicon sequencing also has limitations. First, it may underestimate the diversity in the community given the biases associated with the polymerase chain reaction amplification method. Second, amplicon sequencing can generate wide estimations of diversity. Third, this method cannot provide data related to biological functions encoded in the genome (Refs Reference Sharpton24–Reference Schloss26).
On the contrary, shotgun metagenomic sequencing targets all genomic DNA available in the given sample. The library preparation and workflow are similar to other whole-genome sequencing methods. It enables us to provide a better species- (Ref. Reference Jovel27) and strain-level classification of microorganisms (Ref. Reference Rausch28). However, despite the advantages, this method has several challenges, such as relatively complex data leading to complicated bioinformatics analyses, the presence of a lot of host DNA and contaminant genomes.
Various analyses can be conducted from the OTU data. Diversity analysis is one of the basic analyses for microbiome studies. In general, we recognise two types of diversity, alpha-diversity and beta-diversity. Alpha-diversity is used to analyse the diversity of the samples’ richness and evenness through several parameters: observed species, Chao1 index, Shannon index, Simpson index and Good's coverage (Refs Reference Willis29, Reference Prehn-Kristensen30). Beta-diversity is used to analyse the species complexity of the samples. Diversity analysis is commonly performed using QIIME software (Ref. Reference Navas-Molina31). Principal coordinate analysis is another method used to obtain the principal coordinates and visualise complex data (Ref. Reference Goodrich32).
A study by Johnson et al. highlighted the potential of the high-throughput sequencing of the entire 16S rRNA gene to provide better taxonomic resolution than is achieved by only targeting the hyper-variable regions with a short-read sequencing platform. They performed an in-silico comparison of full and partial 16S rRNA sequencing that showed the importance of utilising the full 16S rRNA gene to accurately classify the microorganisms at high taxonomic resolution (Ref. Reference Johnson33).
NGS can also provide high-throughput transcriptome sequencing or RNA sequencing data. It enables us to detect, quantify and perform deep analyses of the well-known transcripts and novel, unknown transcripts. Although microarrays are also a powerful method to study the transcriptome, RNA sequencing (RNASeq) using NGS offers several advantages. For example, RNASeq can be performed without prior knowledge of the genome sequences, therefore, without reference genomes. RNASeq can also be used to directly measure all RNA transcripts, whereas microarrays are indirect, measuring fluorescence after hybridisation using a probe sequence. Furthermore, RNASeq can be used to detect novel transcripts, and, finally, RNASeq data provide less noise compared with microarrays, in which cross-hybridisation may increase background signals (Ref. Reference Rao34).
The types of sequencers used for NGS should also be considered. Because of the huge number of NGS platforms currently available, choosing the most suitable sequencer is important. This will depend mostly on the study design and the project goal. Other considerations are the run time, read length, number of reads per run, maximum output and price.
Overview of gastric microbiota and factors affecting the composition
The successful identification of Campylobacter pyloridis by Barry Marshal and Robin Warren in the early 1980s was a breakthrough in medicine. Campylobacter pyloridis, renamed Helicobacter pylori in 1984, was shown to successfully colonise the human stomach, which debunked the old ‘sterile stomach’ theory. This discovery suggested the possibility of human stomach colonisation by other pathogenic microorganisms. However, the comprehensive characterisation of gastric microbiota was still not carried out because of the limitations of conventional methods such as culture, histology or immunohistochemistry. The development of NGS methods has enabled better assessment of the composition of stomach microbiota. In addition, most of the early gastric microbiota studies focused on bacteria rather than other microorganisms such as fungi or parasites.
Because of the anatomical location of the stomach, the microbiota detected in the stomach might include microbiota from the oral cavity or duodenum. For example, there is an abundance of Lactobacillus and Veillonella, previously described as oral microbiota (Refs Reference Burcham35, Reference Caufield36). However, these are transient bacteria, and their importance in developing gastric diseases is still not clear.
Bik et al. characterised the microbial diversity within the human stomach using the 16S rDNA clone library. They identified 128 phylotypes and assigned them into five major phyla: Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes and Fusobacteria. Not surprisingly, they found that 67% of phylotypes in the study have been described as phyla commonly found in the oral cavity. They also demonstrated an abundance of transient bacteria (Ref. Reference Bik37). Importantly, the same study showed that H. pylori was the most abundant species in the stomach of H. pylori-infected subjects. A study conducted by Nam et al. on 20 Korean patients reported that gut microbiota differed between subjects from different geographical regions. They proposed that microbiota might be affected by host and environmental factors, such as genetic variations and diet (Ref. Reference Nam38). Five dominant phyla were characterised from the stomachs of Korean subjects: Actinobacteria, Firmicutes, Bacteroidetes, Fusobacteria and Proteobacteria. Another study by Ferreira et al. also supports these results; the gastric microbiota in 135 Portuguese was dominated by five phyla: Proteobacteria (69.3%), Firmicutes (14.7%), Bacteroidetes (9.0%), Actinobacteria (4.3%) and Fusobacteria (1.3%). Therefore, the studies showed no differences in phyla between subjects from different geographical areas, and the differences might be more pronounced at the level of genus, species or subspecies. Delgado et al. examined specimens from 12 healthy subjects from different populations. They found that Streptococcus, Proprionibacterium and Lactobacillus were the most abundant genera, and the microbiota was similar at the phylum level (Ref. Reference Delgado39). The gastric microbiota is more dynamic at the genus level, and various factors might affect this level. Gastric microbiota also reportedly changed with the development of gastric diseases, including H. pylori infection. Studies have investigated the relationship between microbiota composition and the development of various gastric diseases (Table 1).
Table 1. Studies investigating the predominant microbiota in gastric diseases

The human stomach is a complex organ with various mechanisms to maintain a healthy state. Therefore, the exact mechanisms determining the gastric microbiota composition are not clearly understood. However, factors such as dietary habits, and the use of antibiotics, anti-inflammatory drugs, probiotics and proton-pump inhibitors or H2-antagonists, are thought to play important roles. The extensive and long-term use of acid secretion inhibitors such as proton pump inhibitors (PPIs) and H2-antagonists is said to be the most important factor influencing the microbiota composition (Ref. Reference Jackson23). Another proposed mechanism influencing the gastric microbial composition is that PPIs may directly target the bacterial and fungi proton-pump mechanisms of organisms such as H. pylori, Clostridium difficile, Candida albicans and Saccharomyces cerevisiae (Ref. Reference Vesper40). A high pH level may facilitate bacterial overgrowth in the stomach (Refs Reference Vesper40, Reference Theisen41). Subsequently, the change of gastric microbiota composition will also affect the gut microbiota. A study by Imhann et al. revealed that the use of PPI significantly alters gut microbiota, reducing its microbial diversity (Ref. Reference Imhann42). Furthermore, bacterial composition in the PPI user group was clustered differently compared with the non-PPI user group (Ref. Reference Imhann42).
In animal models, dietary habit was reported to be associated with the disturbance of normal gastric microbiota (Ref. Reference He43). Moreover, a long-term high-fat diet was associated with gastric dysbiosis. However, the mechanism leading to dysbiosis in the mouse model might also be caused by the metabolic syndrome, which comprises obesity, hyperlipidaemia and insulin resistance. Probiotics contain live microbial components that have a beneficial effect on human health (Ref. Reference Kechagia44). They have, therefore, been proposed as an alternative or supplemental therapy for gastrointestinal diseases. For example, Igarashi et al. showed the potential of LG21 probiotic to restore the gastric microbiota composition of patients with functional dyspepsia (Ref. Reference Igarashi45). The patients had an alteration of gastric microbiota compared with healthy subjects. In the functional dyspepsia group, the level of Bacteroidetes was higher than that of Proteobacteria, with absent Acidobacteria. In contrast, in healthy subjects, the Proteobacteria level was higher than that of Bacteroidetes, and Acidobacteria were present. The LG21 probiotic supplementation was able to return the gastric microbiota of dyspeptic patients to a composition similar to that of healthy subjects, significantly decreasing the Bacteroidetes/Proteobacteria ratio and increasing Acidobacteria levels. In addition, the abundance of Lactobacillus, which is the genus of the LG21 strain was not increased after 12 weeks therapy (Ref. Reference Igarashi45), showing that the huge amount of LG21 administered, caused a minimum of side effects and no Lactobacillus overgrowth in the stomach.
Overview of gastric transcriptome studies
The metagenomic approach is a powerful method for profiling the gastric microbiota. It is a culture-independent method for characterising microorganisms within the human stomach. It is considered a breakthrough from conventional culture-based or histological approaches, which could only characterise approximately 30% of gastric microbiota. However, metagenomics can only provide information on the ‘presence’ or ‘absence’ of microorganisms; therefore, another approach is needed to shed light on the functional profile of the gastric microbial community. The value of transcriptome studies in examining viable bacteria and microbial gene expression in the human stomach has been shown by Thorell et al. (Ref. Reference Thorell46). Their results showed that H. pylori is the predominant microbiota both in infected individuals and in most uninfected individuals. Conventional methods such as urea breath test, serology and culture confirmed this. H. pylori abundance is positively correlated with the presence of Campylobacter, Deinococcus and Sulphurospirillum. Importantly, the results also showed that the expression of H. pylori genes involved in pH regulation and nickel transport was high (Ref. Reference Thorell46).
Transcriptome studies use a different approach than metagenome studies, focusing on expressed genes in the specific environment (Ref. Reference Aguiar-Pulido47), either from the microbial community or the host. Transcriptome studies analyse gene expression by capturing the mRNA found in a specific environment (Refs Reference Wolf48, Reference Wang, Gerstein and Snyder49), thus determining the viable microbiota. The pipeline for transcriptome studies is shown in Figure 1.
As the mRNA is extracted from biopsy or stool specimens, it may contain microbial, and host RNA; differentiating between the two has proven challenging. A suitable transcriptome reference database should be used to provide accurate data on the expressed genes either from the host or the microbes. Differentiating between microbial and host RNA may help provide sequencing data with less background noise caused by the abundant host RNA. Currently, commercial kits used to separate and isolate bacterial RNA from mixed samples are widely available. Several studies have demonstrated the use of bacterial RNA enhancement kits to clean up samples by removing non-bacterial RNA (Refs Reference Kamminga50–Reference Jorth52).
It is important to note that transcriptome studies can be performed from either the microbial or host perspective. The biological significance depends on the design and aims of the study. For example, where a study aims to examine the expression of genes responsible for antibiotic resistance mechanisms, the microbial transcriptome may provide more significant data. On the contrary, when the study aims to examine the effect of infection on the host, the host transcriptome may give deeper information on the disease pathogenesis. The researcher should consider the best approach to fulfil the study goal.
Microbiome, transcriptome and gastric diseases
The success of H. pylori in colonising the stomach demonstrates the important role of gastric microbiota in gastroduodenal disease pathogenesis. Other microbiota might be able to survive in the extreme gastric niche environment and be related to disease pathogenesis. Therefore, it is important to examine the association between gastric microbiota and various gastric diseases. The microbiome modification during gastric diseases is shown in Table 2.
Table 2. Studies investigating microbiota modification in subjects with diseases

Gastric cancer
Gastric cancer is the fifth most common malignancy globally (age-standardised rates 11.1 per 100 000 in 2018; Global Cancer Observatory). The incidence of gastric cancer shows wide variation worldwide, with a notably high prevalence in East Asian countries such as Japan, Korea, Mongolia and China (Refs Reference Rawla and Barsouk53, 54). H. pylori is the most successful colonising pathogen in the human stomach. It was classified as a class I carcinogen in 1994 by the International Agency for Research on Cancer. H. pylori is one of the most important risk factors for gastric cancer. Several studies have reported a strong association between gastric cancer development and H. pylori infection (Refs Reference Uemura55, Reference Kumar56). Gastric cancer develops and progresses slowly over years, starting from gastric inflammation/gastritis, and progressing through gastric atrophy, intestinal metaplasia, finally developing into gastric cancer. H. pylori plays an important role in inducing gastritis. Moreover, because of H. pylori being able to persistently colonise gastric epithelial cells, chronic inflammation continues in the stomach.
The eradication of H. pylori is crucial for reducing gastric cancer risk. Lee et al. showed that H. pylori eradication prevents gastritis progression into gastric cancer in the INS-GAS mice model (Ref. Reference Lee57). H. pylori infection accelerates the development of premalignant lesions, intestinal metaplasia and severe dysplasia, whereas the administration of antibiotics and eradication therapy as early as 8 weeks post-infection prevents dysplasia, atrophy and intestinal metaplasia (Ref. Reference Lee57). Studies highlighting the significant role of gastric microbiota in gastric disease pathogenesis include another study by Lofgren et al. (Ref. Reference Lofgren58). The absence of gastric colonisation with microbiota in germ-free INS-GAS mice significantly delayed gastric cancer development. In addition, H. pylori alone is capable of promoting gastritis and gastrointestinal intraepithelial neoplasia in INS-GAS mice. Furthermore, H. pylori-infected INS-GAS mice with complex microbiota develop neoplasia faster compared with H. pylori-mono-association INS-GAS mice with the absence of gastric microbiota.
Transcriptome studies can be used to analyse the host genes that might be responsible for gastric cancer. Zhang et al. used transcriptome analysis to identify the genes and pathways involved in gastric adenocarcinoma pathogenesis (Ref. Reference Zhang59). They found that there were 1477 upregulated and 282 downregulated genes in the gastric adenocarcinoma group, compared with the normal controls. Moreover, the functional enrichment analysis and clustering analysis also showed that the upregulated differentially expressed genes (DEGs) were significantly associated with cell adhesion molecule binding, serine hydrolase activity and several inflammation and tumour pathways, such as the p53 pathway, tight junction pathway, apoptosis pathway and tumour necrosis factor signalling pathway. In addition, the relationship of the expression of genes such as RASGRP3 and CTHRC1 to prognosis suggests their potential use as prognostic markers.
Transcriptome studies can also be used to investigate novel therapies for gastric cancer by targeting genes associated with cancer development. For example, Ren et al. found that the GPNMB gene is highly expressed in gastric cancer patients and indicates a worse prognosis. Knockdown of this gene in the gastric cell lines, such as AGS cell and NCI-N87, inhibits the proliferation and migration of cancer cells. Moreover, GPNMB could affect the expression of coinhibitory molecules in the cancer cell and be involved in the escape of cancer cells from the host immune system. Therefore, the GPNMB gene is a promising target for gastric cancer immunotherapy (Ref. Reference Ren60).
Gastritis
Li et al. examined the microbiota composition of healthy subjects and non-H. pylori gastritis patients (Ref. Reference Li61). By using 16S rRNA sequencing, they found that Firmicutes was the most abundant phylum and Streptococcus the most abundant genus in patients with antral gastritis, compared with healthy controls. In addition, there was an under-representation of Proteobacteria in antral gastritis patients. Streptococcus, Prevotella, Porphyromonas, Neisseria and Haemophilus were the most common genera in antral gastritis patients (Ref. Reference Li61).
Nookaew et al., using the microarray method, examined the transcriptomic changes in corpus atrophic gastritis patients facilitated by H. pylori infection (Ref. Reference Nookaew62). They found a significant depletion of DEGs in both antrum and corpus of corpus atrophic gastritis patients. Interestingly, they observed the antralisation process in the corpus atrophy patients, characterised by increased gastrin expression and downregulation of several corpus-specific genes. They also revealed that in corpus atrophy patients, the acidic mammalian chitinase (AMCase) gene had the most significant reduction of expression because of the high expression of this gene in normal corpus mucosa (Ref. Reference Nookaew62).
Metabolome studies complement metagenome and transcriptome studies
Metabolomics is the comprehensive analysis of metabolites from the specimen. The small molecules released by the organisms into the environment are identified and quantified to describe alterations in the environment. The metabolome is considered the most direct indicator of a healthy or altered environment's condition. Variations in the production of signature metabolites are related to changes in the activity of metabolic routes; therefore, metabolomics is useful for pathway analysis (Ref. Reference Gu63). In addition, metabolomics shows promise for use in drug discovery and pharmacogenomics (Refs Reference Wishart64, Reference Tuyiringire65).
Metabolome data are obtained by a very different method compared with microbiome and transcriptome data. The latter data are obtained through nucleotide sequencing, whereas metabolome data are obtained by mass spectrometry, chromatography and nuclear magnetic resonance. Mass spectrometry has been widely used to measure metabolites with high sensitivity. Chromatography is used to separate more complex mixtures of metabolites (Ref. Reference Pan and Raftery66). These methods can be combined as liquid chromatography-mass spectrometry, gas spectrometry-mass spectrometry or capillary electrophoresis-mass spectrometry (CE-MS) to provide reliable data with high precision (Ref. Reference Pan and Raftery66).
A systematic review by Huang et al. analysed 52 molecular epidemiologic metabolomics studies of human upper gastrointestinal cancers (Ref. Reference Huang67). Metabolomic studies have facilitated effective biomarker detection in gastric cancer, supporting the potential of applying metabolomic profiling in cancer prevention and disease management. Although several metabolites have been identified for gastric cancer, the identification of putative metabolomic biomarkers has remained inadequate. Application of metabolomic profiling to molecular epidemiologic studies on gastric cancers may provide insights into the biological significance of crucial metabolites and metabolic pathways, but there is no information on the underlying mechanisms. Given the multi-stage progression of gastric carcinogenesis, metabolic biomarkers associated with precancerous and early gastric cancers must be identified to improve screening and early diagnosis in high-risk populations.
Considering the importance of metabolome studies, it is logical to integrate them with microbiome and transcriptome studies. This addition could provide insights into the outcome of changes in gene expressions, which may lead to differential expression of specific metabolites that impact the health of the host environment. Understanding the whole ecosystem will open exciting approaches for generating new knowledge.
Conclusion and future insights
In future, the integration of multiple datasets obtained from microbiome, transcriptome and metabolome studies will provide solutions to some current challenges. The application of individual methods might not be enough to provide accurate data to support hypotheses because of their limitations. The integration of different studies will allow the researchers to build and test models of microbial activity and inter-microbe or microbe–host interactions. This will enable a better understanding of the association between the environment and the microbial community. For example, the combination of metagenomics and metatranscriptomics may reveal overexpression or underexpression of particular functions and, in some cases, the activities of specific organisms. However, network analysis is crucial for analysing the combination of microbiota, expressed genes and metabolites data obtained with each method to draw accurate conclusions. Only by performing complex network analysis between microbiota, transcriptome and metabolome data we can shed new light on the association between the host, agent and environment.
Acknowledgements
This study was funded by the Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan to YY. LAW and DD were PhD students supported by the Japanese government (MEXT) scholarship program for 2015 and 2016, respectively.
Conflict of interest
The authors declare no competing interests.



