Introduction
Wolbachia is an intracellular alpha-proteobacterium that infects an estimated 66% of arthropod species (Hilgenboecker et al., Reference Hilgenboecker, Hammerstein, Schlattmann, Telschow and Werren2008). Wolbachia are vertically transmitted from parents to offspring only through the egg cytoplasm. In order to enhance maternal transmission, Wolbachia manipulates host reproduction by inducing cytoplasmic incompatibility (CI), male-killing, parthenogenesis and feminization of genetic males (O'Neill et al., Reference O'Neill, Hoffmann and Werren1997).
Previous studies have reported different sequence types (i.e. alleles) for the wsp gene within a host species. Some host species are often detected with two or more alleles of a Wolbachia gene, referred to as multiple infections. Double or triple infection in a single host species are well documented (Breeuwer et al., Reference Breeuwer, Stouthamer, Barns, Pelletier, Weisburg and Werren1992; Vavre et al., Reference Vavre, Fleury, Lepetit, Fouillet and Boulétreau1999; Kondo et al., Reference Kondo, Ijichi, Shimada and Fukatsu2002a). Detection of four or more alleles (especially defined as superinfections here) are reported in a few insects, such as ants, fruit flies and beetles (Malloch et al., Reference Malloch, Fenton and Butcher2000; Jamnongluk et al., Reference Jamnongluk, Kittayapong, Baimai and O'Neill2002; Reuter & Keller, Reference Reuter and Keller2003; Dedeine et al., Reference Dedeine, Ahrens, Calcaterra and Shoemaker2005). Superinfection is caused by frequent occurrence of horizontal transmission of Wolbachia from other host species (Jamnongluk et al., Reference Jamnongluk, Kittayapong, Baimai and O'Neill2002), mutation of Wolbachia genome (Malloch et al., Reference Malloch, Fenton and Butcher2000) and/or recombination between different Wolbachia strains co-existing in a single individual (Reuter & Keller, Reference Reuter and Keller2003). Genetic recombination confounds phylogenetic analyses of Wolbachia based on single loci, e.g. 16S rDNA, ftsZ or wsp (Werren & Bartos, Reference Werren and Bartos2001; Baldo et al., Reference Baldo, Lo and Werren2005). Therefore, Baldo et al. (Reference Baldo, Hotopp, Jolley, Bordenstein, Biber, Choudhury, Hayashi, Maiden, Tettelin and Werren2006) has proposed a multilocus sequence typing (MLST) scheme for Wolbachia, which uses five housekeeping and ubiquitous genes to study Wolbachia strain relationships.
The subfamily Scolytinae (Curculionidae) is composed of subcortical-feeding insects (bark beetles) and fungus-feeding beetles (ambrosia beetles) (Rudinsky, Reference Rudinsky1962). Some of them seriously damage forest trees, and their ecology and evolutionary history have been extensively studied for the purpose of pest control (e.g. Kirkendall, Reference Kirkendall1983, Reference Kirkendall, Wrensch and Ebbert1993; Beaver, Reference Beaver, Wilding, Collins, Hammond and Weber1989; Normark et al., Reference Normark, Jordal and Farrell1999; Farrell et al., Reference Farrell, Sequeira, O'Meara, Normark, Chung and Jordal2001). The alnus ambrosia beetle, Xylosandrus germanus, is one such species. It is highly inbred with female-biased sex ratio (>0.9) (Kaneko, Reference Kaneko1965) that is reduced with outbreeding (Peer & Taborsky, Reference Peer and Taborsky2004). However, outbreeding reduces egg viability (Peer & Taborsky, Reference Peer and Taborsky2005). This observation is suggestive of Wolbachia infections (Peer & Taborsky, Reference Peer and Taborsky2005).
The objectives of this study were to reveal whether Wolbachia infects X. germanus; and, if so, to investigate the Wolbachia infection pattern and evolutionary history in X. germanus, which may provide information on outbreeding depression. We performed amplification and sequence of five MLST and wsp Wolbachia genes and conducted molecular phylogenetic analyses. The revealed pattern of Wolbachia infection was then compared to the X. germanus phylogeny based on mitochondrial COI.
Materials and methods
Insect collection
We collected flying adults of this species at nine sites in Japan in 2005 and 2006 (table S1). Ethanol-bait traps, 10-ml vials filled with 99.5% ethanol (Ito et al., Reference Ito, Kajimura, Hamaguchi, Araya and Lakatos2008), were set up in a mixed stand of broad-leaved trees and shrubs and trapped insects were collected two weeks post set up. Trapped live insects were all females because males of X. germanus are not capable of flying in forests (Kaneko, Reference Kaneko1965). Captured insects were placed in absolute ethanol and stored at −30°C until DNA was extracted. Numbers of samples used in this study are shown in table S1.
DNA extraction and PCR
DNA was extracted from the abdomens of individual specimens. After the abdomens were crushed, each homogenate was incubated with 200 μl of 5% (wt/wt) Chelex-100 sodium (sigma) and 4 μl of 20 mg μl−1 Proteinase K at 56°C overnight. After boiling, the supernatant was used directly as the PCR template.
Because, in this study, we used two distinguishable regions of ftsZ (Holden et al., Reference Holden, Brookfield and Jones1993; Baldo et al., Reference Baldo, Hotopp, Jolley, Bordenstein, Biber, Choudhury, Hayashi, Maiden, Tettelin and Werren2006), we described ftsZ-a and ftsZ-b, respectively (table S2). We amplified three Wolbachia genes (wsp, 16S rDNA and ftsZ-a) and a mitochondrial gene (COI) of X. germanus by PCR using specific primer pairs (table S2). Each 10-μl reaction volume consisted of 1 μl of DNA extract, 0.5 μl of dNTPs (2.5 mM each), 0.05 μl of Taq polymerase (5 U μl−1), 5.45 μl of sterile water, 1 μl of 10× buffer (TAKARA) and 1 μl of forward and reverse primers for the target gene (100 μM). We carried out the standard PCR following conditions: denaturation for 3 min at 94°C, 35 cycles of 94°C for 1 min, the optimal annealing temperature (55°C for wsp, 50°C for ftsZ-a and 16S rDNA, and 48°C for COI) for 1 min and 72°C for 1 min, and final extension at 72°C for 10 min. PCR products were visualized in 1.5% agarose gel under natural light by staining with Mupid Blue (ADVANCE-BIO) or under UV illumination by staining with ethidium bromide.
Cloning and sequencing
PCR products of wsp (550–600 bp) in 3–6 individuals from Furano, Sapporo, Iwate and Aichi, respectively, were cloned with the p-GEMT Easy Vector (Promega) using ampicillin and X-gal blue-white selection system. About ten white colonies expected to contain the inserted plasmid from each product were directly subjected to PCR using the primers M13M4 (5′-GTT TTC CCA GTC ACG AC-3′) and M13RV (5′-CAG GAA ACA GCT ATG AC-3′), useful for determining the length of the inserted DNA fragment. The colonies containing the expected fragment were isolated and cultured in 2 ml of LB medium with ampicillin, and the purified plasmid DNA were directly sequenced using M13M4 and M13RV primers. From the medium, purified plasmids (50 μl) were eluted using a QIAprep-Spin Miniprep Kit (Qiagen). A dye terminator-labeled cycle sequencing reaction was conducted with BigDye DNA Sequencing Kit ver. 3.1 (PE Applied Biosystems). Reaction products were analyzed using an ABI PRISM 310 Genetic Analyzer (PE Applied Biosystems). The temperature profile was 96°C for 10 s followed by 25 cycles of 96°C for 10 s, 50°C for 5 s and 60°C for 4 min. We assigned the wsp alleles in X. germanus the names ‘wXge1-5’, according to wsp sequences.
Amplicons of 16S rDNA, ftsZ-a and COI were purified using QIAprep-Spin Miniprep Kit (Qiagen) and bi-directionally sequenced using BigDye DNA Sequencing Kit ver. 3.1 (PE Applied Biosystems). Reaction products of these genes then were analyzed by the same method as those of wsp.
The wsp, ftsZ-a, 16S rDNA and COI sequences determined were deposited in the DDBJ/EMML/GenBank nucleotide sequence databases. Accession numbers are shown in table S3a, b.
Phylogenetic analysis
Multiple alignments of wsp and COI sequences were conducted using the program package clustalW (Thompson et al., Reference Thompson, Higgins and Gibson1994). The final alignment was inspected and corrected manually using the sequence analysis software BioEdit 7.0.5.3 (Hall, Reference Hall1999). Ambiguously-aligned regions were excluded from phylogenetic analysis. Nucleotide sites, including alignment gaps, were also omitted from the analysis. Phylogenetic trees were constructed with neighbor-joining (NJ), maximum parsimony and UPGMA methods, using the program package mega 3 (Kumar et al., Reference Kumar, Tamura and Nei2004) or PHYLIP 3.65 (Felsenstein, Reference Felsenstein2004). Bootstrap tests were conducted with 1000 resamplings.
For the phylogenetic tree of the COI sequence, we selected only five X. germanus populations (Furano, Sapporo, Iwate, Aichi and Tottori). The other four populations (Yamagata, Saitama, Kochi and Miyazaki) were very similar in haplotypes to Aichi and Tottori (Ito et al., Reference Ito, Kajimura, Hamaguchi, Araya and Lakatos2008). As an outgroup species, we used the closely-related species, Xylosandrus crassiusculus (Motshulsky), captured in the traps placed in Aichi.
Detection of Wolbachia alleles infecting each individual
Wsp alleles in each X. germanus sample were detected by diagnostic PCR. Allele-specific reverse primers (table S4) were designed according to wsp or 16S rDNA sequences. Multiplex-PCR for detection of wXge1, wXge2 and wXge3/wXge5 (lengths of expected PCR products were 312, 245 and 406 bp, respectively) was performed in 20-μl reaction volumes consisting of 1 μl of DNA extract, 1 μl of dNTPs (2.5 mM each), 0.2 μl of Taq polymerase (5 U μl−1), 9.8 μl of sterile water, 2 μl of 10× buffer (TAKARA), 3 μl of wsp81F and 1 μl of each reverse primer (wxge1wr, wxge2wr and wxge3/5wr) (100 μM). wXge4 was detected by standard PCR using another reverse primer (wxge4wr) (255 bp). For wxge3/5wr-positive individuals, standard PCRs for detection of wXge3 and wXge5 were also conducted using different reverse primers, wxge3wr and wxge5_16sr, respectively. The conditions of multiplex PCR and standard PCR for wXge4 detection were identical to those of wsp PCR. To discriminate between wXge3 and wXge5 (489 and 632 bp, respectively), we carried out touchdown PCR using the following conditions: denaturation for 3 min at 94°C; 94°C for 1 min, 70°C decreasing by 2°C for 1 min and 72°C for 1 min; 35 cycles of ten cycles of 94°C for 1 min, 50°C for 1 min and 72°C for 1 min and final extension at 72°C for 10 min. All PCR products were visualized in 1.5% or 2% agarose gel for standard PCR and multiplex PCR, respectively, under natural light by staining with Mupid Blue (ADVANCE-BIO) or under UV illumination by staining with ethidium bromide. To check whether accurate alleles were detected, a few randomly selected PCR products were sequenced directly.
Multilocus sequence typing method for Wolbachia
The MSLT method consists of (i) PCR, (ii) sequenced, (iii) assigned and (iv) reconstruction of the MLST tree. MLST genes (gatB, hcpA, coxA, ftsZ-b, fbpA) were amplified by touchdown PCR using specific primers (table S2). The PCR conditions were as follows: denaturation for 3 min at 94°C; 94°C for 30 s, 70–50°C decreasing by 2°C for 45 s and 72°C for 1 min; 35 cycles of 94°C for 30 s, 50°C for 45 s and 72°C for 1 min and final extension at 72°C for 10 min. PCR products of these genes were purified and bi-directionally sequenced by the same method as 16S rDNA, ftsZ-a and COI. Because MLST genes of wXge4 could not be amplified and sequenced, we removed the strain from MLST analysis.
Wolbachia strains were assigned to their own sequence types (ST), defined as the combination of five alleles in MLST genes. Strain and host information was deposited in the MLST database at http://pubmlst.org/wolbachia (see table S5).
UPGMA and NJ trees were reconstructed from MLST allelic profiles (table S5) using START2 program (Jolley et al., Reference Jolley, Feil, Chan and Maiden2001). Moreover, the concatenated alignment of MLST genes (2079 bp) for the phylogenetic tree was analyzed as described above.
Results
Wolbachia infection in local populations of X. germanus
The wsp gene was successfully amplified from all the specimens analyzed, confirming fixation of Wolbachia infections in all X. germanus populations.
Wolbachia alleles in X. germanus
Five alleles for wsp were detected. Lengths of the five wsp sequences, coded as wXge1to wXge5, ranged from 538 to 580 bp. Based on molecular phylogenetic analysis using wsp sequences, our five wsp sequences were apparently independent of each other (fig. 1).

Fig. 1. Molecular phylogenetic tree of five Wolbachia alleles infecting X. germanus based on the wsp gene.
wXge3 differed from wXge5 by only 2 bp. With respect to ftsZ-a and 16S rDNA, however, genetic differences between wXge3 and wXge5 were greater (ftsZ-a: 4 bp/728 bp; 16S rDNA: 5 bp/853 bp). Therefore, we regarded wXge3 and wXge5 as different alleles and designed primers for detecting infecting Wolbachia based on the polymorphisms existing between these sequences for either the wsp and 16S rDNA (table S4) (see Materials and methods).
Multilocus sequence typing for Wolbachia
We obtained a phylogeny of the five Wolbachia strains using an MLST method based on Wolbachia-housekeeping genes with strains from Baldo & Werren (Reference Baldo and Werren2007) and the MLST database (fig. 2a). Sequence tagging (ST) profiles are shown in table S5. Because MLST genes of wXge4 could not be amplified, we excluded this strain from MLST analysis. wXge1 (ST-131) and wXge2 (ST-138) were closely related to ST-82 and ST-119, respectively (fig. 2a), containing two common alleles (table S5). wXge3 and wXge5 belonged to the ST-139 and ST-140 complex, which shares three alleles with each other (table S5). The complex is phylogenetically similar to the ST-88 and ST-130 complex (fig. 2a).

Fig. 2. (a) MLST tree based on sequence type (ST) profiles and (b) neighbor-joining (NJ) tree based on concatenated alignment of MLST genes of four Wolbachia strains infecting X. germanus without wXge4, including other strains from Baldo et al. (Reference Baldo, Prendini, Corthals and Werren2007) and the database http://www.pubmlst/wolbachia.
A molecular phylogenetic tree using the concatenated alignment of MLST genes (2079 bp) is shown in fig. 2b. All the Wolbachia strains in this analysis belonged to the A supergroup as defined by Werren et al. (Reference Werren, Zhang and Guo1995). wXge1 was closely related to ST-12, ST-66 and ST-73, and wXge2 to ST-65. wXge3 and wXge5 showed the greatest similarity to ST-2, not to ST-88 or ST-130.
Detection rates and polymorphism of Wolbachia
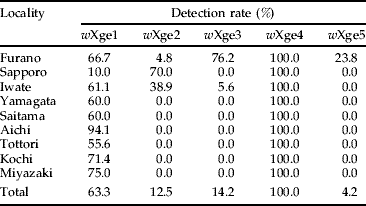
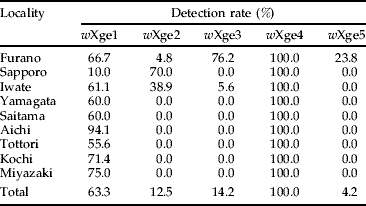
Prevalence of each Wolbachia alleles found by diagnostic PCR is summarized in table 1. wXge4 was detected in all the insects tested. wXge1 also occurred in all the populations accounting for >55% of the infection rates except that from Sapporo. wXge2 was detected in Furano, Sapporo and Iwate, wXge3 in Furano and Iwate and wXge5 only in Furano. The detection rates for these alleles were lower than those for wXge1, except for wXge2 in Sapporo and wXge3 in Furano.
Table 1. Detection rates (%) of each Wolbachia allele in Japanese X. germanus populations.
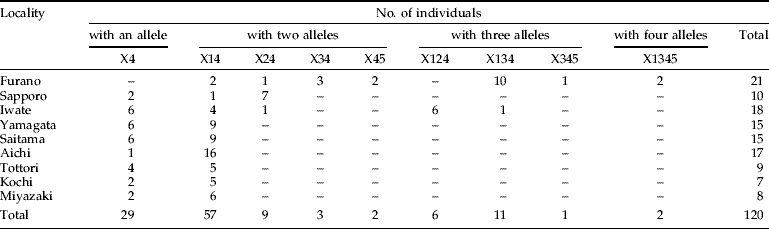
The maximum number of Wolbachia combinations in X. germanus was 16 (=24) because wXge4 was detected from all the individuals. We have developed an abbreviation system for the possible combinations (e.g. detection of three alleles, wXge1, wXge2 and wXge3, is represented as X123). At least nine combinations out of 16 were detected from nine populations (table 2): one pattern of a allele (X4); four combinations of two alleles (X14, X24, X34 and X45); three combinations of three alleles (X124, X134 and X345); and one combination of four alleles (X1345). The Furano population showed the greatest combination of Wolbachia alleles (seven combinations). In Sapporo and Iwate, there were three and five Wolbachia combinations, respectively. In contrast, Yamagata, Saitama, Aichi, Tottori, Kochi and Miyazaki were less polymorphic (X4 and X14).
Table 2. Wolbachia infection polymorphism in Japanese X. germanus populations.
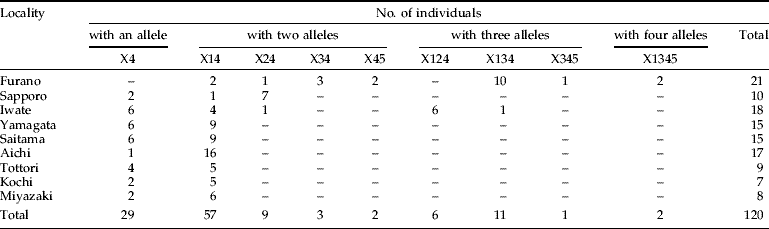
Combinations of Wolbachia alleles are shown in abbreviations (e.g. triple detection of wXge1, wXge2 and wXge3 as X123).
Relationship of Wolbachia combinations with X. germanus haplotypes
A molecular phylogenetic tree based on COI of mtDNA in X. germanus is shown in fig. 3 (cf. table S6). Description of haplotypes (Xg2–36) and clades (A–C) was determined according to Ito et al. (Reference Ito, Kajimura, Hamaguchi, Araya and Lakatos2008). Six new haplotypes (Xg31–36) were found in this study. Clade A (Xg02–04, 10, 31, 35 and 36; N=40) was consistently found in all five populations, whereas clade B (Xg22–24, 27, 32–34; N=17) belonged to individuals from Furano and Iwate. Clade C (only Xg30; N=3) was found only in Furano.

Fig. 3. Molecular phylogenetic tree of X. germanus haplotypes based on CO1 of mtDNA with reference to Wolbachia combinations.
Figure 3 also illustrates relationship between Wolbachia combination and COI haplotypes. Individuals in clade A had wXge1 and/or wXge2 in addition to wXge4 (X4, X14, X24 and X124). On the other hand, wXge2 was absent from Clades B and C: X14, X34, X45, X134, X345 and X1345.
Discussion
Wolbachia infection in an ambrosia beetle
This is the first report of Wolbachia infection in ambrosia beetles. Wolbachia infections had already been found in four species of bark beetles: Ips typographus (Stauffer et al., Reference Stauffer, van Meer and Riegler1997), Hypothenemus hampei (Vega et al., Reference Vega, Benavides, Stuart and O'Neill2002), Coccotrypes dactyliperda (Zchori-Fein et al., Reference Zchori-Fein, Borad and Harari2006) and Pityogenes chalcographus (Arthofer et al., in press). However, there are no sequence data of Wolbachia infecting I. typographus, C. dactyliperda and P. chalcographus, and the data for H. hampei (accession number: AF389084) is too short to compare with our wsp sequences.
In X. germanus, outbreeding depression reduces egg viability (Peer & Taborsky, Reference Peer and Taborsky2005), suggesting CI induced by Wolbachia. In our results (figs 1 and 2), X. germanus was determined to be infected with at least five Wolbachia strains and with a total of nine allele combinations (table 2). Although we did not investigate Wolbachia phenotypes in the present study, our results imply outbreeding depression is probably caused by Wolbachia-induced multi-directional CI, which occurs in crosses where both males and females are infected with different CI-inducing Wolbachia (e.g. Hoffmann & Turelli Reference Hoffmann, Turelli, O'Neill, Hoffmann and Werren1997). We recognize the possibility that female-biased sex ratios in X. germanus may be caused by infection of other sex-altering bacteria (e.g. Cardinium: Gotoh et al., Reference Gotoh, Noda and Ito2007). Surveys of such bacteria are planned in the future.
Comparison of MLST analysis with wsp-based phylogeny
In the present study, we determined Wolbachia taxonomy by both wsp-based phylogeny (fig. 1) and MLST analysis (fig. 2). Although molecular phylogenetic analysis, based on wsp, has often been reported since Zhou et al. (Reference Zhou, Rousset and O'Neil1998), the surface protein wsp is highly recombinant and wsp-based inferences are not reliable (Baldo & Werren, Reference Baldo and Werren2007). After the MLST method was proposed as the better analysis for Wolbachia taxonomy (Baldo et al., Reference Baldo, Hotopp, Jolley, Bordenstein, Biber, Choudhury, Hayashi, Maiden, Tettelin and Werren2006), some studies using MLST have been carried out (Baldo & Werren, Reference Baldo and Werren2007; Baldo et al., Reference Baldo, Prendini, Corthals and Werren2007, Reference Baldo, Ayoub, Hayashi, Russell, Stahlhut and Werren2008; Zabalou et al., Reference Zabalou, Apostolaki, Pattas, Veneti, Paraskevopoulos, Livadaras, Markakis, Brissac, Mercot and Bourtzis2008; Narita et al., Reference Narita, Shimajiri and Nomura2009; Ratchoudhury et al., Reference Raychoudhury, Baldo, Oliveira and Werren2009 (cf. MLST database)). However, the MLST tree (fig. 2a) has low resolution in the point of phylogeny because it focused on differentiation of each-gene sequences not sequences themselves (Maiden et al., Reference Maiden, Bygraves, Feil, Morelli, Russell, Urwin, Zhang, Zhou, Zurth, Caugant, Feavers, Achtman and Spratt1998). Therefore, at the moment, molecular phylogenetic analysis using concatenated alignment of MLST genes may reveal Wolbachia evolution more clearly.
Historical dynamics of Wolbachia strains infecting X. germanus
We have detected five alleles of wsp gene from X. germanus. Detection of distinct wsp alleles in a single specimen is common (Breeuwer et al., Reference Breeuwer, Stouthamer, Barns, Pelletier, Weisburg and Werren1992; Vavre et al., Reference Vavre, Fleury, Lepetit, Fouillet and Boulétreau1999; Kondo et al., Reference Kondo, Ijichi, Shimada and Fukatsu2002a). However, only four studies reported detection of more than five alleles in one insect species: ants Formica exsecta (five alleles: Reuter & Keller, Reference Reuter and Keller2003), Solenopsis daguerrei (nine alleles: Dedeine et al., Reference Dedeine, Ahrens, Calcaterra and Shoemaker2005), the fruit fly Bactrocera ascita (five alleles: Jamnongluk et al., Reference Jamnongluk, Kittayapong, Baimai and O'Neill2002) and the raspberry beetle Byturus tomentosus (seven alleles: Malloch et al., Reference Malloch, Fenton and Butcher2000).
wXge4 is unique because only wsp gene was amplified in all the insects tested. Some recent papers have reported lateral gene transfer from Wolbachia to host insects (Kondo et al., Reference Kondo, Nikoh, Ijichi, Shimada and Fukatsu2002b; Hotopp et al., Reference Hotopp, Clark, Oliveira, Foster, Fischer, Torres, Giebel, Kumar, Ishmael, Wang, Ingram, Nene, Shepard, Tomkins, Richards, Spiro, Ghedin, Slatko, Tettelin and Werren2007). Although we do not have any direct evidence, wXge4 might be the only gene fragment inserted in the host chromosome.
The phylogenetic analyses (figs 1 and 2) show that Wolbachia infecting X. germanus are divergent between wXge3 and wXge5; the difference in the wsp sequence between wXge3 and wXge5 was only 2 bp, and wXge3 was identical in three out of five MLST genes with wXge5 (cf. table S5). These results suggest that a common strain differentiated to wXge3 or wXge5. Although when they were differentiated has not been clear, it may have occurred in the current host, X. germanus.
We have found nine different combinations with four Wolbachia alleles, without wXge4, in X. germanus (table 2). To date, this species has the most variable combinations of Wolbachia among the reported hosts. This combination may have been formed by multiple horizontal transmissions to different host lineages. wXge2 and wXge3/wXge5 did not infect the same individuals (table 2) and their combinations were related to the beetle phylogeny (fig. 3). Ito et al. (Reference Ito, Kajimura, Hamaguchi, Araya and Lakatos2008) implied that X. germanus has already developed into three lineages (clades A, B and C) before colonization of Japan. Thus, wXge1 and wXge4 may infect the common ancestor of three clades of X. germanus before its differentiation. After that, wXge2 and the ancestor strain of wXge3 and wXge5 may infect its descendants, clades A and B/C. Finally, wXge3 and wXge5 would differentiate from the common Wolbachia.
Acknowledgements
We thank A. Davies, T. Hagimori-Adachi, T. Kikuchi and Y. Oba for providing technical advices and information on Wolbachia. We also thank L. Baldo, F.E. Vega and H. Anbutsu for their critical reading of the manuscript. We are grateful to the following people for collecting the beetle specimens: K. Iguchi, A. Ueda, S. Saito, K. Nakamura, K. Ishida, M. Inoue, S. Sato and K. Araya.
This study was supported by Grants-in-Aid for Scientific Research from JSPS (18380090, 18405012, 20405025), the Fujiwara Natural History Foundation (2004), the Inamori Foundation (2005), the IFO Foundation (Institute for Fermentation, Osaka) (2007), and the Shouwahoukoukai Foundation (Ito Chube'e) (2008).
Supplementary material
The following online table can be viewed at http://journals.cambridge.org/ber:
Table S1. Location of sampling sites and numbers of individuals tested in each year.
Table S2. Information on primers for amplifying Wolbachia and host insect X. germanus DNA.
Table S3a. Accession numbers of three genes (wsp, ftsZ-a and 16S rDNA) of each Wolbachia strain.
Table S3b. Accession numbers of COI of mtDNA in each haplotypes of X. germanus and X. crassiusculus.
Table S4. Information on reverse primers for differentiating between Wolbachia alleles.
Table S5. Sequence type profile of each Wolbachia strain used in MLST.
Table S6. Composition of X. germanus haplotype on COI of mtDNA in each locality.