Introduction
Cryopreservation of oocytes and embryos is a crucial step for the widespread conservation of animal genetic resources. Currently, there are two techniques for gamete and embryo cryopreservation: slow freezing and vitrification. Vitrification has become a viable and promising alternative to traditional approaches, especially when dealing with in vitro-produced or micromanipulated embryos and oocytes (Pereira & Marques, Reference Pereira and Marques2008) or transgenic embryos (Chrenek et al., Reference Chrenek, Bauer and Makarevich2011, Reference Chrenek, Turanová, Slamečka and Makarevich2013).
Many variables in the vitrification process, including the type and concentration of cryoprotectant, the temperature of the vitrification solution at exposure, the type of device that is used for vitrification, the species and genotype of the animal, the developmental stage of the embryos, as well as the type (in vivo versus in vitro) of embryo production, can profoundly influence its effectiveness (Vajta & Kuwayama, Reference Vajta and Kuwayama2006). The two most important parameters for the success of vitrification are the rates of the cooling and warming (Morato et al., Reference Morato, Izquierdo, Paramio and Mogas2008) and the effects of dissolved substances, especially the concentration of the cryoprotectants (Almasi-Turk et al., Reference Almasi-Turk, Roozbahi, Alibadi, Haeri, Sadeghi and Hosseni2009).
Minimizing the toxicity of the cryoprotectant can be achieved through the additional use of non-permeable polymers such as polyvinylpyrrolidone (PVP), polyethyleneglycol (PEG), or Ficoll 70. In addition, the toxicity of the cryoprotectant can be lowered by using a combination of two cryoprotectants and a stepwise exposure of cells to pre-cooled concentrated solutions (Orief et al., Reference Orief, Schultze-Mosgau, Dafopoulos and Al-Hasani2005).
The efficiency of the vitrification method can be estimated in various ways, such as the post-thaw survival of embryos in culture, stereomicroscopical evaluation of morphology or ultrastructural evaluation of morphology using transmission electron microscopy (TEM). Cuello et al. (Reference Cuello, Bethelot, Delaleu, Venturi, Pastor and Vazquez2007) demonstrated that vitrified-warmed porcine blastocyst hatching during in vitro culture appears to coincide with good ultrastructure and low cell-death index, suggesting that the hatching rate assessed by stereomicroscopy is a more appropriate parameter than embryo re-explantation for an evaluation of the quality of vitrified-warmed blastocyst. Ultrastructural studies carried out with vitrified bovine embryos (Vajta et al., Reference Vajta, Hyttel and Callesen1997) have shown that certain abnormalities and alterations remain undetected by stereomicroscopy only. A study by Bettencourt et al. (Reference Bettencourt, Bettencourt, Silva, Ferreira, de Matos, Oliveira, Ramao, Rocha and Sousa2009) revealed that the extension of ultrastructural damage, especially of mitochondria and cytoskeleton, was related to the light microscopy semithin evaluation but not to stereomicroscopical embryo scoring at thawing. This finding suggests that embryo scoring according to semithin evaluation is a useful tool for the prediction of ultrastructural lesions. Although, the studies focused on bovines (Fair et al., Reference Fair, Lonergan, Dinnyes, Cottel, Hyttel and Ward2001), porcine (Fabian et al., Reference Fabian, Gjorret, Berthelot, Martinat-Botté and Maddox-Hyttel2005; Cuello et al., Reference Cuello, Bethelot, Delaleu, Venturi, Pastor and Vazquez2007), ovine (Bettencourt et al., Reference Bettencourt, Bettencourt, Silva, Ferreira, de Matos, Oliveira, Ramao, Rocha and Sousa2009) and rabbit (Popelkova et al., Reference Popelkova, Chrenek, Pivko, Makarevich, Kubovicova and Kacmarik2005) embryo ultrastructure findings after vitrification were published, the ultrastructural changes of transgenic rabbit embryos after vitrification have not been documented to date.
The aim of this study was to evaluate the efficiency of ethylene glycol, Ficoll 70 and sucrose (EFS) vitrification solution for vitrification of rabbit gene-microinjected embryos at the morula stage, and to characterize the ultrastructural alterations induced in gene-microinjected enhanced green fluorescent protein (EGFP)-positive (EGFP+) and EGFP-negative (EGFP–) embryos by the vitrification process.
Materials and methods
Egg collection
New Zealand White rabbit does (4–6-month-old) reared on the local farm were superovulated using a single intramuscular (i.m.) injection of 20 IU/kg pregnant mare's serum gonadotrophin (PMSG; Werfaser, Alvetra & Werfft, Vienna, Austria), 72 h before mating. Immediately prior to mating with a male of proven fertility, the females were injected i.m. with human chorionic gonadotrophin (hCG; Werfachor, Alvetra & Werfft) at 40 IU/kg live weight. The pronuclear stage eggs were flushed from the oviduct of slaughtered animals with phosphate-buffered saline (PBS) supplemented with 5% fetal calf serum (FCS) both purchased from Gibco BRL (Auckland, New Zealand) 19–20 hours post coitum (hpc). The flushed eggs were evaluated morphologically and the eggs with two pronuclei, two polar bodies and compact cytoplasm were selected and randomly divided into two groups: (1) intended for gene microinjection; and (2) non-microinjected (non-Mi, intact) eggs.
Gene microinjection
Gene microinjection was carried out using an Olympus microscope equipped with micromanipulation units (Alcatel, France) and a microinjector (Eppendorf, Germany). The DNA solution (EGFP; Clontech, USA) was injected into the male pronucleus of rabbit eggs in CO2-independent medium (CIM; Gibco BRL) supplemented with 10% FCS (Chrenek et al., Reference Chrenek, Bauer and Makarevich2011, Reference Chrenek, Turanová, Slamečka and Makarevich2013). Eggs in the untreated, non-Mi group were transferred into four-well dishes (Nunc, Roskilde, Denmark) that contained 600 μl of knockout Dulbecco's Modified Eagle's Medium (k-DMEM; Gibco BRL) supplemented with 10% FCS immediately after flushing, whilst the other group was subjected first to microinjection followed by a transfer of Mi eggs into culture medium. Embryos were incubated for up to 72 hpc (morula stage) at 39 °C in a humidified atmosphere of 5% CO2 in air up to vitrification process.
Vitrification protocol
CO2-independent medium supplemented with 20% FCS (Gibco BRL) was used as a basic solvent for the vitrification solution composed of ethylene glycol (EG, 40%), Ficoll 70 (18%) and sucrose (0.3 M) (EFS medium; Kasai et al., Reference Kasai, Hamaguchi, Zhu, Miyake, Sakurai and Machida1992); dilution medium was 0.5 M sucrose. Vitrification of embryos at morula stage was performed as described previously (Kasai et al., Reference Kasai, Hamaguchi, Zhu, Miyake, Sakurai and Machida1992; Papis et al., Reference Papis, Sypecka, Korwin-Kossakowski, Wenta-Muchalska and Bilska2005, Chrenek et al., Reference Chrenek, Turanová, Slamečka and Makarevich2013). Briefly, five to eight embryos were subjected to equilibration in EFS solution for 3–4 min at room temperature (22 ± 1 °C). Within this period of time, embryos were mixed with the vitrification solution by repeated aspiration into glass capillaries and then were inserted into a middle column of EFS solution in a preloaded, 0.25 ml insemination straw (Minitübe, Germany) in a small volume (<0.5 μl) of medium. The straw was kept above the surface of the liquid nitrogen for 60 s before being plunged into it. After 1–3 days of embryo storage, straws were warmed in air (10 s) and the contents of each straw were expelled into 2 ml of 0.5 M sucrose solution. At approximately 7–8 min later, embryos were transfered to CIM/FCS to complete the EG dilution. Subsequently, a proportion of the warmed morulae (EGFP+, EGFP–) was fixed and taken for electron microscopy analysis. The control group consisted of intact non-microinjected and non-vitrified morulas. The remainder of the warmed morulas were cultured additionally for 3 days in k-DMEM medium supplemented with 10% FCS. In vitro development of embryos to the blastocyst stage within 48 h and to the hatching/hatched blastocyst stage within 72 h was recorded.
Electron microscopy
Embryos were assigned into the following groups: (1) vitrified microinjected EGFP+ embryos; and (2) microinjected EGFP– embryos. The control group consisted of intact non-microinjected and non-vitrified embryos. Embryos were placed into Karnovsky's fixative (2% paraformaldehyde and 2.5% glutaraldehyde in 0.15 M cacodylate buffer, pH 7.1–7.3) for 1 h at 4 °C, and then washed twice in cacodylate buffer for 5 min (Karnovsky, Reference Karnovsky1965). Subsequently, the embryos were embedded into 4% agar individually and postfixed in 1% osmium tetroxide in cacodylate buffer for 1 h. Samples were then dehydrated by passage through an ethanol series, embedded into Durcupan ACM (Fluka), and serially sectioned into ultrathin sections. Ultrathin sections were collected on copper and nickel grids, contrasted with uranyl acetate and lead citrate and examined under a transmission electron microscope (JEM 100 CXIl, Jeol, Japan) operating at 80 kV.
Morphometry
The volume density of cellular components was determined using the point-count method (Weibel & Bolender, Reference Weibel, Bolender and Hayat1973). Briefly, a transparent grid that consisted of 154 total test points was laid over each micrograph. The square occupied by each component was expressed as a percentage of the number of points that covered the structural component relative to the total number of test points of the test grid. Within an individual embryo, the average volume density represents total counts from the micrographs of the whole embryo.
Statistical analysis
The data on volume density (%) of cellular components were expressed as mean ± standard error of mean (SEM) and assessed using analysis of variance (ANOVA) and the Kruskal–Wallis test. Effect of treatment was considered to be statistically different at a P-value <0.05.
Results
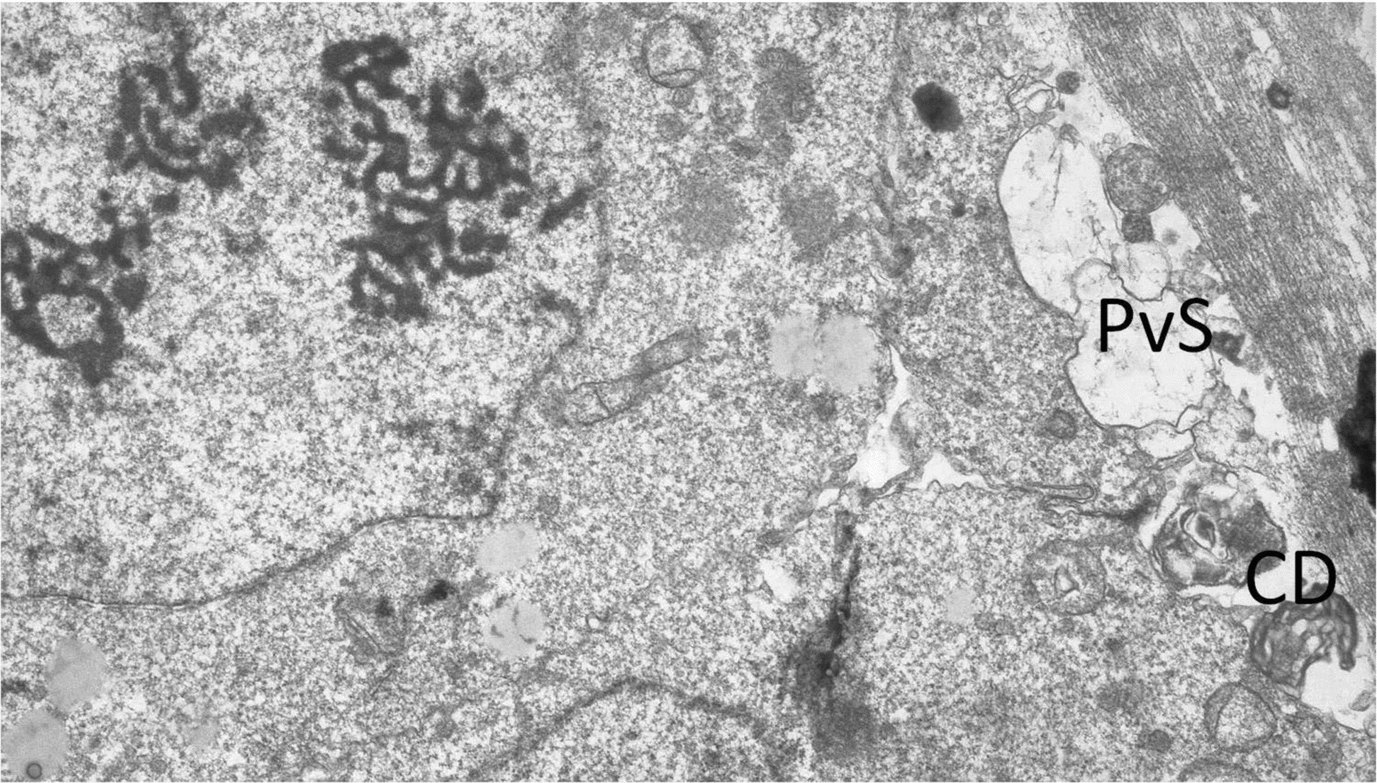
We found a higher content of cellular debris (4.02%; Fig. 1) and flocculent vesicles (6.82%) in vitrified EGFP– embryos in contrast with the non-vitrified control (Table 1). In vitrified EGFP+ embryos only the fat droplets content (11.60%) was increased. The proportion of dense bodies present in the cells of experimental and control embryos appeared to be similar (5.16% and 4.76% versus 3.88%, respectively), a finding that suggested that vitrification does not affect this ultrastructure parameter significantly.
Table 1 Changes in volume density (%) of undamaged cellular components of devitrified and control rabbit embryos (mean ± standard error of the mean (SEM))

n, Number of random micrographs.
a ,b Within rows, values with different superscripts differ at P < 0.05 (one-way ANOVA; Kruskal–Wallis test).
EGFP, enhanced green fluorescent protein.
Figure 1 Higher content of cellular debris (CD) and flocculent vesicles in vitrified enhanced green fluorescent protein (EGFP)-negative (EGFP–) embryos in the perivitelline space (PvS), magnification ×7200.
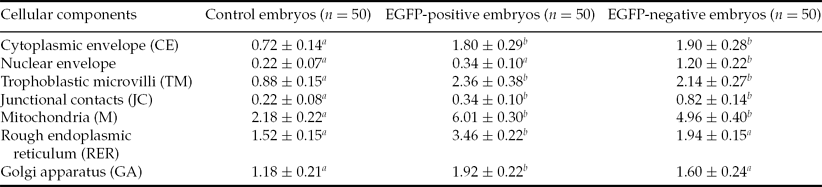
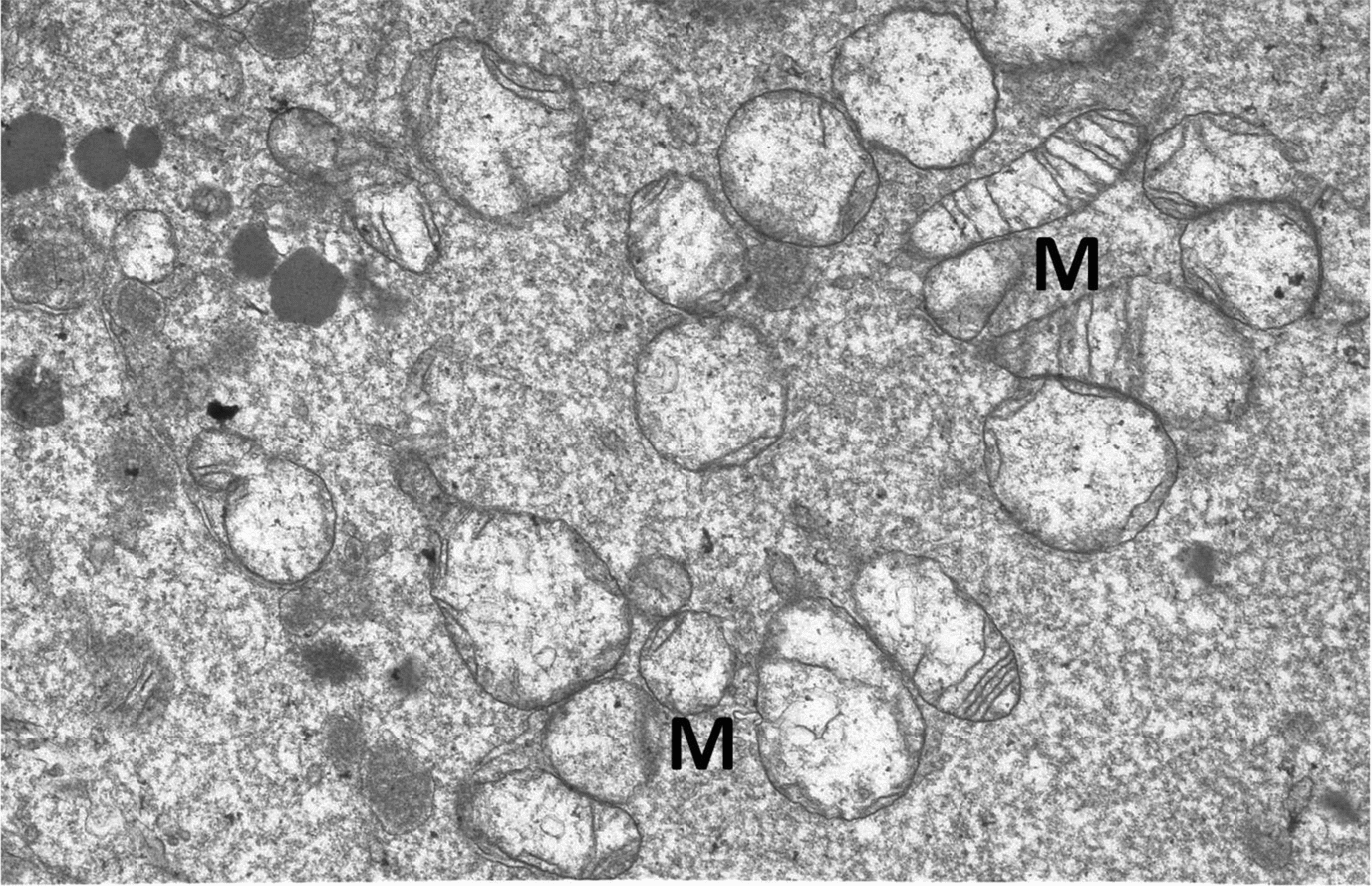
Major organelles damaged in the process of vitrification in both EGFP+ and EGFP– vitrified embryos compared with the control embryos were mitochondria (6.01% and 4.96% versus 2.18%, Fig. 2), trophoblastic microvilli (2.36% and 2.14% versus 0.88%), plasma membrane (1.80% and 1.90% versus 0.72%) and rough endoplasmic reticulum (3.46% and 1.94% versus 1.52%, respectively; Table 2). A higher proportion of deteriorated cell structures and organelles may be caused by the vitrification process rather than by mechanical violation (by the gene-microinjection procedure), as a detailed inspection of the ultrastructure revealed that most damage occurred in the membrane structures.
Table 2 Changes in volume density (%) of damaged cellular components of devitrified and control rabbit embryos (mean ± standard error of the mean (SEM))
n, Number of random micrographs.
a ,b Within rows, values with different superscripts differ at P < 0.05 (one-way ANOVA; Kruskal–Wallis test).
EGFP, enhanced green fluorescent protein.
Figure 2 Major organelles (M, mitochondrion) damaged in the process of vitrification in both enhanced green fluorescent protein (EGFP)-positive (EGFP+) and EGFP-negative (EGFP–) vitrified embryos, magnification ×7200.
Discussion
Vitrification has achieved wide attention for use with embryos from several mammalian species. This technique should allow the storage of embryos for extended periods with reduced loss of their capacity to develop further into live offspring. Li et al. (Reference Li, Lai, Wax, Hao, Murphy and Rieke2006) have reported the birth of transgenic pigs after in vitro production and OPS vitrification. This report shows the potential of vitrification for practical application.
Dimethylsulfoxide (DMSO), propylene glycol (PG), glycerol, or ethylene glycol (EG), alone or as mixtures, have been used as cryoprotective additives for vitrification of rabbit embryos. Cryoprotectant mixtures (e.g. EG and DMSO) may have some advantages over solutions that contain only one penetrating cryoprotectant. However, the overall composition of the solution is also important, as embryo and oocyte survival is modified by other components such as sugars, macromolecules, or polymers, and salts. High concentrations of polymers such as Ficoll and dextran appear to be non-toxic to the embryos and can be used to replace an approximately equal amount of EG without changing the solution glass transition temperature (Visintin et al., Reference Visintin, Martins, Bevilacqua, Mello, Nicácio and Assumpção2002). Recently, Makarevich et al. (Reference Makarevich, Chrenek, Olexikova, Popelkova, Turanova, Ostro and Pivko2008) documented significant higher survival rate, lower apoptotic index and better actin filament quality for the vitrified rabbit embryos at morula stage when using EFS-based solution in comparison with equi-molar concentrations of EG plus DMSO.
In the present study the vitrification solution (EFS) composed of 40% EG, 18% (v/w) Ficoll 70 and 0.3 M sucrose was used for vitrification in straws. Devitrified gene-microinjected embryos showed a high survival rate evaluated on the basis of number of cells to reach blastocyst (90%) or hatching stages (86%) (data not shown). In the control group all embryos reached the blastocyst stage and 98% of them were further maintained to cleave into hatching blastocyst stage. This difference was observed to be not significant. The study of Papis et al. (Reference Papis, Sypecka, Korwin-Kossakowski, Wenta-Muchalska and Bilska2005) was conducted using a similar experimental outline (the same vitrification protocol and embryo developmental stage). Among all treated embryos, 71% were able to develop into the blastocyst stage, but only 23.5% developed to term after embryo transfer. Papis et al. used a different genotype of the embryo donors (paralytic tremor (pt) rabbit mutant lane of the Chinchilla breed), which could be a reason for the lower efficiency of the vitrification method. It seems that pt rabbit embryos are more sensitive and have lower cryoresistance. As was found in former studies, some differences can be observed in embryo development and quality in terms of embryo freezability among different strains of mice (Rall et al., Reference Rall, Schmidt, Lin, Brown, Ward and Hansen2000), as well as in rabbits (Vicente & Garcia-Ximenez, Reference Vicente and Garcia-Ximenez1993).
Abe et al. (Reference Abe, Hara, Matsumoto, Kobayashi, Sasada, Ekwall, Rodriguez-Martinez and Sato2005) compared a single and stepwise exposure of bovine cumulus oocyte complexes to the solution, which was composed of 40% EG, 18% (w/v) Ficoll-70 and 0.3 M sucrose (EFS40) using a nylon-mesh holder. In the stepwise exposure, few abnormalities were observed compared with the single-step exposure, in which most oocytes showed a highly vacuolated cytoplasm with many ruptured mitochondria. Investigations by Boonkusol et al. (Reference Boonkusol, Faisaikarm, Dinnyes and Kitiyanant2007) were focused on developmental capacity and ultrastructural changes of matured buffalo oocytes vitrified using 35 or 40% EG as vitrification solution for solid surface vitrification (SSV) and in-straw vitrification (ISV). Both methods of vitrification caused profound ultrastructural modifications to microvilli, mitochondria, oolemma and cortical granules, however damaged mitochondria were more abundant in ISV vitrified oocytes than in SSV vitrified oocytes, a finding that correlated with developmental data. Bettencourt et al. (Reference Bettencourt, Bettencourt, Silva, Ferreira, de Matos, Oliveira, Ramao, Rocha and Sousa2009) assumed that cryopreservation methods must be chosen according to the species and embryonic stage and improvements in cryopreservation and thawing methods to decrease cellular damage of embryos are still warranted.
Toxicity of high concentrations of cryoprotectants can be attributed to osmotic stress and biochemical injuries, which depend on the rate of permeation and concentration of cryoprotectants and is influenced by the duration and temperature of exposure. Osmotic damage results from important changes in volume during addition and dilution of cryoprotectants. Intrinsic biochemical toxicity may result from specific interaction between cryoprotectant and cell components (e.g. proteins, DNA and biochemical membranes) or from non-specific effects of cryoprotectants on the environment of cellular biomolecules (e.g. ionic strenght, pH, surface tension, and redox potential). The early injuries, such as loss of microvilli, intercellular junctions and cytoplasmic membrane stability, could have a disruptive impact over the cytoskeleton, which would then lead to a severely disturbed cell organization, such as that found in degenerated blastomeres (Kaidi et al., Reference Kaidi, Van Langendonckt, Massip, Dessy and Donnay1999). We have found significantly higher changes in volume densities of cytoplasmic envelope, trophoblastic microvilli and junctional contacts in both vitrified groups (EGFP+ and EGFP–) compared with the non-vitrified control. However, we can suggest that these structural changes, observed by TEM immediately after thawing, were not detrimental for subsequent developmental competence of Mi embryos, and can be restored during further embryo culture. This suggestion is based on a high developmental competence and hatching rates of devitrified Mi-derived embryos documented in post-culture.
In both vitrified groups (EGFP+ and EGFP–) cell damage was associated with mitochondrial injuries that ranged from severe matrix swelling with loss of cristae to membrane degeneration and rupture. Damaged mitochondria after cryopreservation has been reported in previous studies (Cocero et al., Reference Cocero, Díaz de la Espina and Aquilar2002; Cuello et al., Reference Cuello, Bethelot, Delaleu, Venturi, Pastor and Vazquez2007), suggesting that the preservation level of the mitochondrial matrix and envelope can be used as a reliable sign of cellular damage. An early loss of the mitochondrial structure leads to disruption of energy supply to cell boundaries. Clear signs of regeneration were observed at the ultrastructural level after in vitro culture periods (for 4 or 24 h after thawing), including the re-establishment of junctional contacts between trophectoderm cells and normalization of the mitochondrial morphology (Vajta et al., Reference Vajta, Hyttel and Callesen1997; Fabian et al., Reference Fabian, Gjorret, Berthelot, Martinat-Botté and Maddox-Hyttel2005; Popelkova et al., Reference Popelkova, Chrenek, Pivko, Makarevich, Kubovicova and Kacmarik2005).
In conclusion, this study describes, for the first time, the ultrastructural evaluation of vitrified rabbit transgenic embryos. The ultrastructural lesions found for EGFP+ and EGFP– embryos vitrified by EFS vitrification solution were similar. Observation of TEM showed slight accumulation of cellular debris and lipid droplets compared with control intact embryos. More severe alteration were detected in membrane structures, such as mitochondria and the cytoskeleton, which can influence vital cell functioning and thus compromise embryo viability after cryopreservation. We suggest that the higher proportion of deteriorated cell structures and organelles may be caused as a consequence of the vitrification process rather than by mechanical violation, as a detailed inspection of ultrastructure revealed most damage in cell membrane structures.
Acknowledgements
This work was supported by the Slovak Research and Development Agency under contracts No. APVV LPP-0119–09 and No. APVV-0556–011, and by the Ministry of Education (Slovak Republic) under contracts KEGA No. 012UPJS-4/2011 and VEGA No. 1/0415/12, and international project A Collaborative European Network on Rabbit Genome Biology (COST-RGB-NET). The research leading to these results has also received funding from the European Community under Project No. 26220220180: Building Research Centre ‘AgroBioTech’. The authors thank Dr Shubhadeep Roychoudhury, Assistant Professor, Assam University, India for language corrections.