I. Background
A. Paradigm Shift in Health Research
Health research in the United States is undergoing a paradigm shift: broadening the ranks of health researchers and expanding the methods of health research. Part of this expansion involves the growth of “big data,” the gathering and analyzing of vast troves of information about large numbers of people as a means to understand population health. Big data health research is an orienting focus of major federal and private research initiatives, including the Precision Medicine Initiative,1 the Electronic Medical Records and Genomics Network,2 and the Personal Genome Project.Reference Angrist3 This research often includes significant public engagement and greater involvement of human research participants, including an active role in planning and conducting the research itself.Reference Woolley and Rothstein4
Big data and an expanded public role in research are cornerstones of citizen science, which may be defined as “a range of participatory models for involving non-professionals as collaborators in scientific research.”Reference Wiggins, Wilbanks, Guerrini, Riesch and Potter5 Citizen science includes enlisting non-experts in the collection, reporting, and analysis of health-related data; expanding health research from its traditional university- or industry-based settings through non-expert or public involvement in the conduct and governance of research; and crowdsourcing research to address specific population or community health needs.Reference Hoffman, Khatib, Irwin, Wright and McGowan6
The growth of nontraditional health research sometimes blurs the line between professional and citizen science.Reference Evans, Fiske, Prainsack and Buyx7 Nontraditional health researchers include, for example, independent researchers, citizen scientists, patient-directed researchers, do-it-yourself (DIY) researchers, and self-experimenters, and it is likely that new research arrangements and activities will be developed in the future.Reference Rothstein, Wilbanks and Brothers8
Several factors help to explain the appeal and growth of these new forms of health research.
Many people are critical of traditional health research, which they view as slow, expensive, unresponsive, and dominated by commercial interests.Reference Berkelman, Li, Gross, Trouiller, Walsh, Grant and Coleman9
Crowdsourcing, N of 1 studies, and other alternative research methodologies have been excluded from traditional research funding mechanisms.Reference Swan10
The popularity and growth of social media and online patient communities facilitates collaboration, recruitment, participation, and dissemination of results.
The growth of DIY culture and the availability of direct-to-consumer (DTC) health-related (including genomic) testing has encouraged research by nonprofessionals.Reference Beresford, Seyfried, Pei and Schmidt11
Public familiarity with digital health data and research platforms have demystified health research and made it seem similar to other forms of data analysis and consumer health technologies.
Smartphones with health apps and other mobile technologies have led to the collection of vast amounts of biometric data.
The potential of unregulated health research using mobile devices was on display in a groundbreaking study of Parkinson’s disease in 2015. To take advantage of new software supporting health research on mobile devices, Sage Bionetworks, an independent nonprofit research organization based in Seattle,12 conducted the first major smartphone-based health research study. The Parkinson’s disease mPower study recruited participants online in partnership with collaborating Parkinson’s disease organizations. The study used a novel, highly visual, self-guided, online consent process. Study data were generated by using the smartphone to record the voices of the participants, their posture and stability, their reaction time, and other measures of symptoms of Parkinson’s disease. Approximately 17,000 participants, an unprecedented number, enrolled in the study over a six-month period. Although this study was not federally funded or otherwise subject to regulation, including IRB review, the study protocol was submitted and approved by the WIRB-Copernicus Group, an independent IRB.
This article reports the results of and builds upon a three-year study at the intersection of health research using mobile devices and unregulated health research. The use of mobile devices in health research has increased significantly, and it is generally subject to the same regulations as other health research conducted by entities covered by the federal research regulations. Unregulated health research uses various methods, including crowdsourcing information, DIY research, and N of 1 studies, which are beyond traditional health research. This article focuses on the intersection of these two important trends.
The new paradigm for health research challenges the regulatory requirements and research ethics norms that govern traditional approaches to human subject protections.13 This new paradigm creates new tensions for research ethics and policy by disrupting the conventional definitions of health experts, funders, researchers, participants, and research settings. A fundamental question is whether it makes sense to apply a single set of regulations to research ethics with widely varying origins, methods, and funding sources.Reference Resnik14 Despite such challenges, the potential value of new research methods was recognized by Congress when it enacted the Crowdsourcing and Citizen Science Act of 2017.15 That act grants federal agencies the authority to use crowdsourcing and citizen science in their research. In addition, the 21st Century Cures Act16 directs the Food and Drug Administration (FDA) to create a trial framework to use “real world evidence” in its medical device oversight.Reference Sherman17
This article reports the results of and builds upon a three-year study at the intersection of health research using mobile devices and unregulated health research.Reference Roth-stein and Wilbanks18 The use of mobile devices in health research has increased significantly, and it is generally subject to the same regulations as other health research conducted by entities covered by the federal research regulations.19 Unregulated health research uses various methods, including crowdsourcing information, DIY research, and N of 1 studies, which are beyond traditional health research. This article focuses on the intersection of these two important trends. (See Figure 1.)
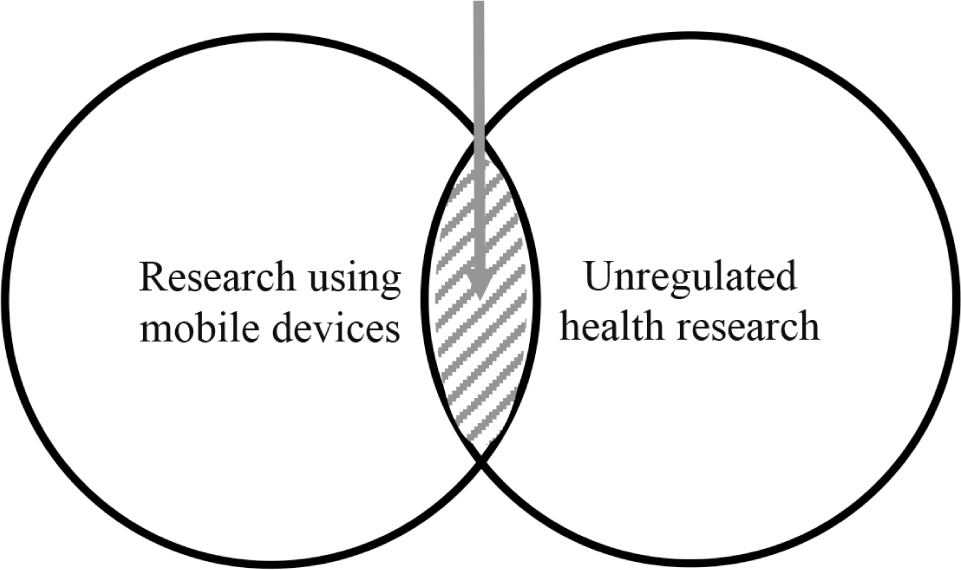
Figure 1. Unregulated Health Research Using Mobile Devices
Mobile devices and their health apps create new research risks based on significantly increased scale. By utilizing DTC genetic testing, publically accessible data repositories, biometric data collection and analysis, and other methods unregulated researchers can produce large-scale studies that raise concerns about balancing risks and benefits, informed consent, privacy, and other issues. Although these are traditional matters for researchers, participants, funders, and IRBs, unregulated health researchers largely operate by their own rules. This article reviews the benefits, risks, and policy alternatives to unregulated health research using mobile devices.
B. Unregulated Health Research and Researchers
The new health research described above is generally “unregulated” and conducted by “unregulated researchers.”Reference Doerr and Guerrini20 As used in this article, “unregulated” means not subject to the federal regulations for the protection of human research subjects adopted by 16 federal departments and agencies (“Common Rule”)Reference Meyer21 or promulgated by the Food and Drug Administration (FDA).22 The research and researchers defined as “unregulated” may still be subject to other federal regulations. These include regulations on unfair or deceptive trade practices enforced by the Federal Trade Commission (FTC),23 or the privacy, security, and breach notification rules promulgated under the Health Insurance Portability and Accountability Act (HIPAA)24 and the Health Information Technology for Economic and Clinical Health Act (HITECH Act).25 “Unregulated” research and researchers also may be subject to regulation under state research laws or other state legislation.26
The definition of research used in this article follows the Common Rule definition, which provides in relevant part: “Research means a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge.”27 Similarly, the definition of “human subject” (or “participant” in this article) follows the Common Rule definition, which provides in relevant part: “Human subject means a living individual about whom an investigator (whether professional or student) conducting research: “(i) Obtains information or biospecimens through intervention or interaction with the individual, and uses, studies, or analyzes the information or biospecimens; or (ii) Obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens.”28 Although research with deidentified specimens or data is not considered human subjects research under the Common Rule, our focus on unregulated research does not make such a distinction.
This article analyzes a wide range of unregulated health research with human participants, but it focuses on nontraditional and emerging types of researchers, such as independent researchers, citizen scientists, patient-directed researchers, and DIY researchers.29 The context is health research using mobile devices. The article does not consider all possible unregulated researchers, such as corporations or foundations. Nor does it consider all forms of unregulated research that can affect human health, including environmental research,Reference Dickinson, Zuckerberg and Bonter30 citizen science gamification,Reference Kreitmair and Magnus31 and citizen scientists and biohackers engaging in interventional clinical research and self-experimentation.Reference Pauwels, Denton and Kuiken32 Although these issues are important, they are beyond the scope of this project.
C. Mobile Devices
The 2007 introduction of the iPhone permanently altered the long-term research landscape. It represented the replacement of devices that merely made phone calls with fully functional pocket computers that also made phone calls. The iPhone and all the other smartphones provided a design into which an increasing amount of hardware could be integrated, emulated in software, or attached to the phone via an app and network. According to recent estimates, 81% of North American adults own a smartphone,Reference Sim33 and more than half of smartphone users are collecting “health-associated information” on their smart-phones.Reference Edwards34 There are more than 325,000 mobile health apps available from the Apple App Store, Google Play Store, and other sources.Reference Pohl and Andrews35
Because of its direct connectivity, the smartphone creates unique research opportunities, such as the ability to pull and push information directly to and from the end user. It also allows developers to simplify complex processes, such as authorization to transfer data stored in online portals to secondary locations. Through ever-expanding hardware, smartphones increasingly contain sensors that can be repurposed from their initial use to surveil populations (e.g., GPS coordinates) and measure at least some elements of health (e.g., accelerometers). And via its connection to secondary mobile devices that tether with apps, a smartphone forms an expandable platform to connect app-navigated health devices and plug-ins, such as glucose meters, electrocardiograms, ultrasound, pulse oximetry, and heart rate monitors.Reference Nebeker36
Mobile devices with biometric measuring capabilities have obvious research implications. Smartphones facilitate access to hundreds of millions of potential research participants in the United States alone and can increasingly measure those participants in highly granular and personal ways. In 2015, Apple accelerated the role of smartphones in research by releasing an open source toolkit called ResearchKit, which makes it easy to build mobile research applications.Reference Chu and Majumdar37 A robust debate on the ethical, legal, and social implications of ResearchKit began shortly thereafter.Reference Duhaime-Ross38 A companion open source Android toolkit called ResearchStack was released in 2016. No clear count of mobile research apps exists, but based on known implementations, more than thirty studies were launched in the first year of ResearchKit alone,Reference Tourraine39 and a similar number of consents for mobile health apps were studied by the Global Alliance for Genomics and Health.Reference Moore40
Mobile devices intersect with a broader consumer technology market built on the integration of persistent behavior surveillance with advertising, with the smartphone deeply connected to the relevant business models.Reference Naughton41 Their apps represent controllable ways to interact with and monitor users. These apps can exploit many features of the phone in ways that consumers may not anticipate, like, or even understand.Reference Gray42 In addition, the numerous uses of smartphones often create an information overload for consumers in looking at terms of service and privacy policies,Reference Obar and Oeldorf-Hirsch43 which are often written at levels that can baffle even graduate-level readers.Reference Litman-Navarro44 The common response of clicking “I agree” without carefully reading (or reading at all) the terms of service or privacy policies raises the issue of whether the consumer has provided valid consent to the uses described in these documents.Reference McDonald and Cranor45
Like other dominant sectors in consumer technology, mobile devices are characterized by a software monopoly. As of 2019, the vast majority of U.S. residents with smartphones used either Google or Apple app stores, with estimates as high as 99.74% using one of the two based on their choice of mobile operating system.46 These two companies decide what apps are appropriate to maintain in their app stores, dictate requirements on what apps must and must not display to consumers, and act to review and take down apps that violate their guidelines. They also possess the power to make or break an app based on, for example, how that app appears in searches, by placement on a featured page, or location within the app store.Reference Vaidhyanathan47
Apple and Google differ in hardware, however, as only an Apple phone runs an Apple operating system, and vice versa, whereas Google’s Android is present on a dizzying variety of handsets. This allows Apple far more control over their hardware ecosystem. Apple leverages this to promote some privacy-supporting features;Reference Jorgensen48 Google’s deep connection to advertising revenue means Android is by default less protective of privacy.Reference Fowler49 They also differ in their approach to app stores; whereas Apple enforces a variety of community norms, Google takes a broad hands-off governance role.50
Apple and Google currently leverage existing software standards, such as SMART (Substitutable Medical Applications, Reusable Technologies) on FHIR (Fast Health Interoperability Resources), to enable access to health records by any healthcare provider with a compatible EHR system;Reference Terry51 and the SMART on FHIR stack is broadly adopted by the emergent health data app community. Efforts around “consumer-led data transfer,” such as the CARIN Alliance, have promulgated forms of normative community regulation through codes of conduct that can also be leveraged in regulatory thinking.52
D. Use Cases
As noted above, this analysis is designed to address scenarios that involve health research being conducted by unregulated researchers via mobile devices. This scope potentially includes a wide variety of scenarios, although there is no formal typology available to organize and reference specific types of scenarios. For this reason we have identified a set of use cases that will help ground the sections that follow, including the discussion of ethical considerations (Part II) and the related policy recommendations (Part III). For definitional purposes, there are at least two characteristics of any study involving unregulated mobile health research: (1) whether the mobile health data were originally collected explicitly for research use, and (2) whether the data are being used for research purposes by the original collector/holder of the data.
The first factor is important because it determines whether app users are likely to understand that they are participants in a research study and not just users of an app. The fact that an app was created explicitly for a research purpose is also important because it points to the intention of the app developer or the organization funding the development of the app to serve as a researcher, thereby presenting additional expectations, such as informed consent. The second factor, whether an app is being used for research by the original collector, is important because the original collector of the data (typically the developer or funder of the app) has had direct contact with the app users and could thus be held responsible for ensuring the participant is informed of the research, has had an opportunity to give affirmative consent, etc. When research is conducted by a secondary recipient of the data, the lack of direct contact between the researcher and the participant means that the researcher is constrained in their ability to directly obtain consent, etc. Another important difference between the original data collected and the secondary recipient is that app users are typically not aware the secondary recipient has acquired their data.
These two criteria, original purpose of data collection and proximity of the research to the participant, can be used to form a 2 x 2 table (Table 1) containing four representative scenarios involving unregulated mobile health research. Although these four cells represent the general use cases, it is important to examine a range of additional characteristics or variations of the four scenarios that are also relevant to the ethical analyses and policy considerations addressed in this article.
Table 1. Framework for Categorizing Unregulated Mobile Health Research
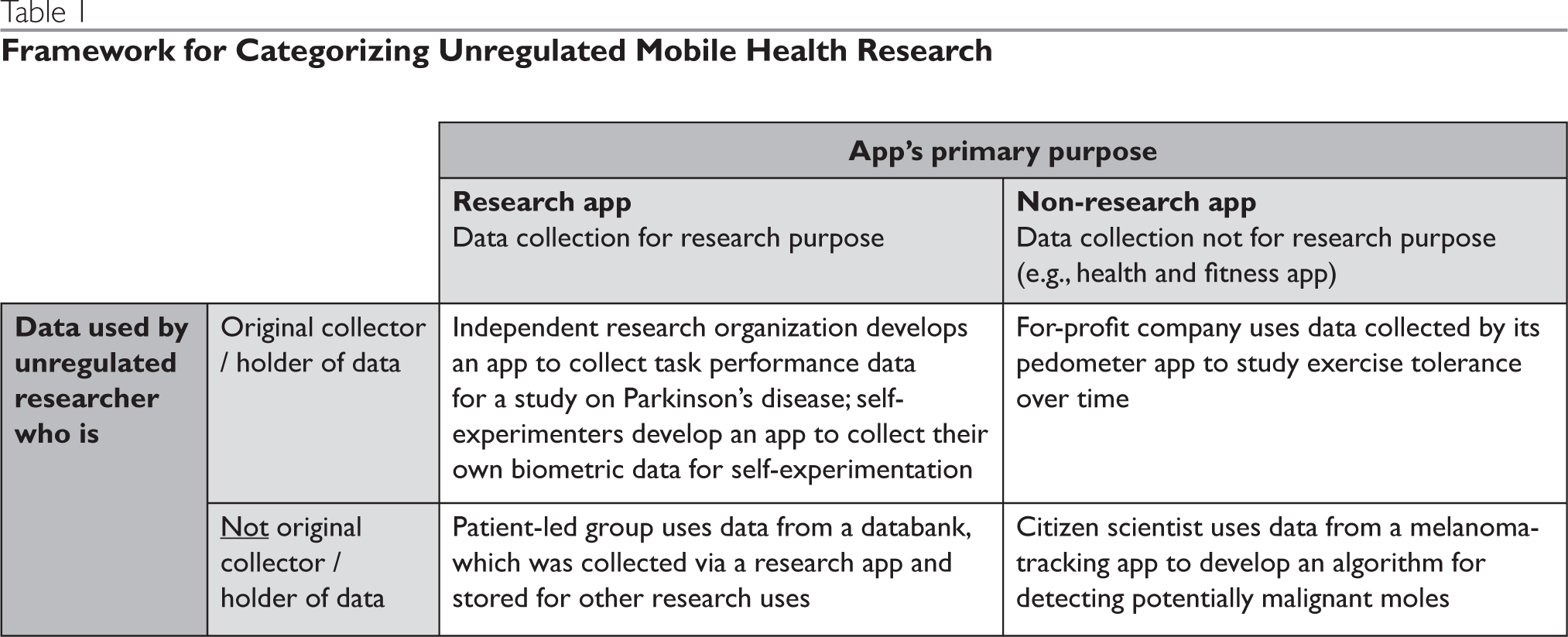
First, there are numerous types of unregulated researchers, including for-profit commercial companies, independent research organizations (i.e., not affiliated with academic or other institutions, such as Sage Bionetworks), independent app developers, individual community/citizen scientists, patient-led groups (e.g., PatientsLikeMe), and individual and group self-experimenters (e.g., Crohnology).
Second, app users may include children or other individuals who lack capacity to make decisions regarding research participation and data inclusion. This presents additional issues, such as verifying the identity of the app user to ensure that any necessary consent is valid.Reference Brothers, Clayton and Goldenberg53
Third, data used in unregulated research vary in sensitivity. For example, GPS data may increase the risk that de-identified data could be re-identified, whereas data relating to certain behaviors or health conditions may be stigmatizing. The combination of different types of data, both public and app-/research-generated, may further increase its sensitivity.
Fourth, an app’s design may or may not provide for return of research results or health-related information. Unregulated research may generate novel individual findings that unregulated researchers may want to return to participants either through the app or by re-identifying app users. This situation raises a variety of issues including the quality and validity of the findings provided, the scientific rigor, validity of data and quality, app users’ expectations and understanding of the limitation of these findings and their privacy interests. Return of results is a complicated matter discussed separately in this symposium.Reference Wolf54
E. Quality Issues
Some unregulated health research using mobile devices satisfies even the most exacting methodological standards; other unregulated research raises serious concerns. Quality issues commonly discussed in the literature include poor study design, use of health apps that convey erroneous health information or inaccurately record biometric data, insufficiently rigorous data analysis, and publication of conclusions beyond what the study supports.Reference Bonney, Fiske, Heyen, Dickel and Brüninghaus55
The lack of scientific rigor in unregulated health research has significant consequences. A poorly designed study raises ethical issues because even minimal risks are not justified.Reference Emanuel, Wendler, Grady, Hood and Flores56 Flawed research may create serious risks to participants and society, as discussed below.57 Also, low quality unregulated mobile health research may tend to discredit all similar research.
One methodological area of concern is the selection and utilization of participants for inclusion in unregulated research.Reference Largent, Lynch and McCoy58 Much of the recruitment and conduct of the research takes place on the internet, and such methods have been criticized as placing too great a reliance on self-recruitment and web-based tools that produce inadequately sized or convenience samples.Reference Janssens and Kraft59 The proportion of highly educated, digitally literate, and well-off people who take part in internet-based research raises concerns about the representativeness of the participants.Reference Del Savio, Prainsack, Buyx, Callier and Fullerton60 Self-reporting of symptoms presents other issues,Reference Vayena and Tasioulas61 including whether participants have been trained adequately to observe conditions and record health data.Reference Resnik62
The methodological concerns raised by some types of unregulated health research strongly suggest that humility and setting “modest goals” are important. Such an approach necessitates “an acknowledgement that methodological questions regarding data quality are still in need of addressing and addressing convincingly, as well as an acknowledgement about the limits of what can be expected from public expertise and contributions.”Reference Riesch and Potter63
The expected increase in unregulated health research using mobile devices strongly suggests that the risk of app-based harms from health research is also likely to grow. Many of the risks described below stem from poor quality in research design, data capture, or analysis. The risk of harm to individuals and groups is mainly in the following four broad categories.
F. Risk of Harms
The lack of legal regulation of certain health research using mobile devices would not be a concern if participants were not placed at risk. Unfortunately, some mobile device and app-based health research poses a significant risk of harm. To date, most of the reported incidents of harm from the use of health apps on mobile devices do not involve research. But, research uses of mobile health apps raise similar issues as health surveillance or wellness uses. Examples include incorrect information and inaccurate measuring, leading to actions or decisions that are adverse to health and wellbeing. The expected increase in unregulated health research using mobile devices strongly suggests that the risk of app-based harms from health research is also likely to grow.64 Many of the risks described below stem from poor quality in research design, data capture, or analysis. The risk of harm to individuals and groupsReference Yu and Juengst65 is mainly in the following four broad categories.
Physical and psychological harms can result when apps used in mobile health research provide erroneous health information that participants rely upon to their detriment. These include improperly diagnosing a condition, recommending that the individual forgo essential treatment or medications, or advising the individual to take harmful or ineffective doses of medications or supplements. In one example, the leading app for managing and diagnosing skin cancer correctly classified just 10 of 93 biopsy-proven melanomas.Reference Feraro, Morrell and Burkhart66 In another example, a systematic assessment of 46 smartphone apps for calculating insulin dose based on planned carbohydrate intake, found that 67% of the apps miscalculated dose recommendations, which put users at risk of poor glucose control or catastrophic overdosing.Reference Huckvale67 In 2019, the FDA warned patients and healthcare providers of the risks associated with unap-proved or unauthorized devices for diabetes management, including glucose monitoring systems, insulin pumps, and automated insulin dosing systems.68
In some cases, harm relates not to the accuracy of the health app, but its use. In one study, some individuals with insomnia who used sleep trackers to improve their sleep became so obsessed with the data produced by the trackers during their sleep that their insomnia worsened, a condition known as orthosomnia.Reference Baron and Chen69 Other tracking apps used by consumers have caused similar harms. For example, orthorexia nervosa is obsessive behavior in pursuit of a healthy diet, which is associated with the use of Instagram, a photo and video-sharing social networking platform.Reference Turner and Lefevre70 Calorie tracking apps have led to disordered eating caused by self-imposed dietary interventions.Reference Simpson and Mazzeo71 Whether a health app is used in a research or a consumer setting, individuals may be harmed if not adequately informed of the psychological as well as physical risks associated with its use.
Dignitary harms, including invasion of privacy and harm to one’s reputation, can result from insufficient privacy protections that lead to the disclosure or sale of sensitive information.Reference Wakefield72 For example, in an assessment of the 36 top-ranked apps for depression and smoking cessation, 29 transmitted data for advertising and marketing purposes to Google and Facebook, but only 12 of 28 transmitting data to Google and 6 of 12 transmitting data to Facebook disclosed this fact.Reference Huckvale, Torous and Larsen73 In a study of 211 Android diabetes apps, permissions required to download the app authorized collection of tracking information (17.5%), activating the camera (11.4%), activating the microphone (3.8%), and modifying or deleting information (64.0%).Reference Blenner74
Mental health data is especially sensitive, and individuals who use mental health apps are likely to be especially vulnerable and perhaps not as attuned to the privacy risks as they ought to be. One app for monitoring people with bipolar disorder and schizophrenia is reported to be “so precise it can track when a patient steps outside for a cigarette break or starts a romantic relationship — and where that new partner lives.”Reference Smith75 Although this app is used in academic research (and presumably regulated), other mental health apps used in other settings are already being marketed for depression, anxiety, PTSD, and other conditions.76 These practices raise three concerns: (1) adequacy of disclosure regarding generation and use of the individual’s information; (2) adequacy of informed consent that uses click-through agreement to download mental health apps; and (3) the possible invasion of privacy of other individuals identified by geolocation features of the app.
Economic harms can result from medical identity theft and other harms caused by inadequate data security or access to an individual’s personal information. For example, apps can access a mobile device user’s contacts, text messages, photos and videos, credit card information, and facial features,Reference McDonald, McDonnell and Mitchell77 thereby facilitating identity theft. In 2017, there were 1,579 data breach incidents, exposing nearly 158 million Social Security numbers, although it is not known how many of these resulted from health apps.Reference Schaffer78 Inadequate security, however, is a well-documented problem with mobile health apps.Reference Dongjing79
Societal harms can result in one of two ways. First, socially-identifiable groups or communities may be harmed when questionable research conclusions lead to increased levels of stigmatization or discrimination. Second, improperly designed or performed research can lead to erroneous scientific conclusions that are detrimentally relied upon by numerous individuals — a societal response to the physical and psychological harms mentioned above. Unregulated health research differs widely in its aims, methods, and quality. As with any research, one must assume that some percentage of unregulated health research is poorly designed or performed.Reference Janssens, Kraft, Fiske, Heyen, Dickel and Bruninghaus80 Unlike regulated research, however, unregulated research has few checks on scientific rigor, such as an IRB considering whether there is a favorable risk-benefit ratio, grant funders evaluating the scientific merits of a proposal, or a peer reviewed journal evaluating the data analysis.Reference Majumder and McGuire81 Consequently, erroneous findings of the research can be widely disseminated over the internet through social networks and other platforms where significant numbers of individuals could learn of and be harmed by a study’s scientifically unsound conclusions.82 Even retracted and repudiated research can thrive on the internet and cause serious harms around the world, as evidenced by the “scientific” articles supporting the anti-vaccination movement Reference Armstrong, Naylor and Kata83 and articles advocating harmful self-help measures to treat cancer and autism.Reference Hauser and Zadrozny84
II. Ethical Considerations
A. Introduction
Unlike many other countries, in the United States the laws and regulations pertaining to research with human participants is highly fragmented, which results in notorious gaps in coverage.85 Whether there is any regulation and, if so, the nature of the regulation depends on the funding source, the identifiability of the specimens and data, and the existence of any applicable state law. Regardless of these differences in legal status and applicable rules, regulated and unregulated research share a common ethical imperative to engage in sound scientific inquiry without undue risk of harm to participants in the conduct of the research and to society in the determination and dissemination of research findings. In Part II, we explore the common ethical foundations of regulated and unregulated health research, and consider them in the context of research using mobile devices and health apps.
This exploration begins by considering the normative grounding for research with mobile devices and health apps. For regulated researchers, their compliance obligations are already prescribed in detail by applicable laws, although the context of mobile devices and health apps presents some novel challenges. Besides legal obligations, many traditional researchers, such as academic medical centers, do not want to violate the trust of their patient communities or the shared commitment to ethical conduct of their professional staff. For unregulated health researchers, the focus of our study, it may be more difficult to satisfy the following, often-conflicting goals inherent in all health research. The primary goal is to safeguard the autonomy, privacy, and other welfare interests of research participants.Reference Rasmussen86 A secondary goal is to minimize the burdens on citizen scientists, health app developers, and other unregulated researchers to preserve their flexibility and capacity to innovate. To point the way for achieving these goals we have endeavored to identify essential ethical principles and best practices that should apply to all research, regardless of the current legal regime.Reference Resnik87
B. Balancing Risks and Benefits
A basic principle of research ethics is that all researchers are ethically obligated to minimize the risks and maximize the potential benefits of research participation.88 Even though research via a mobile health app typically does not involve invasive testing or medical interventions, it nonetheless exposes participants to risks including physical harms, dignitary and psychological harms, economic harms, and societal harms. Unregulated researchers have the same ethical obligation as other researchers to minimize the above-noted harms to individuals who participate in their research. At a minimum, this means assessing the potential risks to participants in these four areas and identifying strategies to minimize any identified risks. Although specific risks will vary from study to study, minimizing the risks of research via mobile health apps generally means using a rigorous study design, transmitting the least amount of identifiable and/or sensitive data needed to achieve the aims of the study, using stringent criteria for quality when selecting health results or advice that will be provided to participants, and reminding users that health apps are no substitute for appropriate, individualized medical care.
While it is fairly intuitive that the risks of research should be minimized, it is far less clear how investigators are expected to maximize the benefits of research.Reference King89 Interventional research with human participants is based conceptually on the idea of equipoise — that the research is being conducted because it is truly unknown whether a new intervention or product (like a wearable) truly provides benefits, whether these benefits outweigh its risks, and whether the balance of risks and benefits are superior to some relevant alternative.Reference Miller and Weijer90 If these things were already known, then research is unnecessary (and any risks created by the research are ethically unjustified). In most cases, the obligation to maximize the benefits of research simply means that research should be conducted in such a way that participants are not precluded from receiving the benefits of interventions that are already known to work. Consider, for example, an app that is designed to use wearable data to inform a user’s workout plan. Even if the developers of the app would eventually want to test whether the app could provide benefits to users in the absence of a personal trainer, they could maximize the benefits to participants by first conducting research with participants who are also receiving the benefits of work with a personal trainer. Only once research of this type had established the benefits and risks of the app in this context would research be conducted to compare the app and a personal trainer head-to-head.
Another threat to an appropriate balance of risks and benefits in unregulated research is the enthusiasm of researchers about the potential of the product, like a new wearable or app they are testing. For example, the recruitment materials for a study may make implicit or explicit representations about the benefits of a new wearable when in fact the research is being meant to determine whether the wearable is, in fact, safe and effective. This confusion about equipoise is problematic not only because it may prevent a participant from appropriately considering the risks and benefits of research participation, but also because research based on the assumption that an intervention or product is beneficial is vulnerable to confirmation bias.Reference Nickerson91
For these reasons, all researchers, including unregulated researchers, should work to suspend their enthusiasm for a new intervention or product when they are conducting research. As much as possible, research should be designed and conducted from a perspective of equipoise. The obligation to maximize the benefits of research should instead be regarded as an obligation to conduct research in the most rigorous way possible so that future users and society as a whole can benefit from the generalized knowledge gained by conducting the research, such as establishing whether a new wearable or app is safe and effective.
Because researchers may be too invested in the success of a study to assess its risks and benefits objectively, it is important for strategies to minimize risks and maximize benefits to undergo review by an individual or entity that is independent from the researcher and is not invested in the outcome of the research. This is discussed in greater detail in section II-F.
C. Consent/Permission
Informed consent has long been considered a cornerstone of research ethics. It is a fundamental demonstration of the ethical principle of respect for persons92 and, with few exceptions, is required for traditionally regulated research. Federal regulations set forth specific elements of information that must be disclosed to prospective participants, as well as the conditions under which consent is obtained.93 Even so, informed consent often fails to achieve its goal of adequately informing participants of key study elements. A substantial body of empirical research has documented problems with consent form length and reading complexity.Reference Albala, Doyle, Appelbaum, Paasche-Orlow, Larson, Foe and Lally94 Further, individual-level risk factors, such as low literacy, low educational attainment, and lack of English fluency (for studies conducted primarily in English),Reference Montalvo and Larson95 may hinder comprehension. Interventions to improve consent comprehension have met with only limited success, although systematic reviews of such studies highlight methodologic challenges.Reference Nishimura and Tamariz96
The movement of research into mobile app forms creates at least two new problems. First, as noted elsewhere in this article, mobile platforms can remove many of the regulatory obligations to obtain informed consent by facilitating research outside the traditional institutions to which regulations normally attach. Second, developers and researchers who voluntarily integrate an informed consent process face barriers related to the specific interaction of mobile devices and comprehension.
A range of approaches has been suggested for informing app/device users about research use of their data,Reference Hammack-Aviran, Brelsford and Beskow97 many of which do not constitute informed consent. For example, “general notification” is an approach involving a brief, broad disclosure that data could be used for research, but offering users no choice in the matter. “Broad permission” similarly involves a brief disclosure, but allows users a simple yes/no choice. Although these kinds of models have some advantages (e.g., low burden, efficiency for research), there are significant concerns that they provide too little detail; users are likely simply to click through such disclosures without reading them, and those who do read them may not fully grasp or remember them.98 Approaches that could meet ethical and regulatory requirements for informed consent include broad consent, categorical or “tiered” consent, and consent for each specific research use. Each of these also entails important advantages and disadvantages from both the user and researcher perspective, many of which have been echoed in other research arenas such as biobanking.Reference Garrison, Steinsbekk, Myskja and Solberg99
Regardless of the approach chosen, key design principles include simple language,Reference Ridpath, Greene and Wiese100 integration of visual elements (e.g., photos, drawings), combined with teach-back approaches.101 Further, experts have suggested including design features that would require some increased attention or additional action by app users in response to research-related disclosures.102 Sage Bionetworks has released a series of toolkits103 and papersReference Doerr104 related to e-consent, facilitating the implementation of best practices by app developers.Reference Wilbanks, Cummings, Rowbotham, McConnell and Ashley105
D. Privacy and Security
Privacy and security are fundamental aspects of the ethical conduct of research involving human participants. Adopted by the World Medical Association (WMA) in 1964, the Declaration of Helsinki establishes a duty of physicians involved in medical research to protect “privacy…and confidentiality of personal information of research subjects.”106 Consistent with the mandate of the WMA, the Declaration of Helsinki is addressed primarily to physician-researchers,107 but it also “encourages others who are involved in medical research involving human subjects to adopt these principles.”108
First prepared by the Council for International Organizations of Medical Sciences in collaboration with the World Health Organization in 1982, the International Ethical Guidelines for Health-Related Research Involving Humans (International Ethical Guidelines) address the use of “data obtained from the online environment and digital tools.”109 In particular, the current (2016) International Ethical Guidelines provide:
When researchers use the online environment and digital tools to obtain data for health-related research they should use privacy-protective measures to protect individuals from the possibility that their personal information is directly revealed or otherwise inferred when datasets are published, shared, combined or linked. Researchers should assess the privacy risks of their research, mitigate these risks as much as possible and describe the remaining risks in the research protocol. They should anticipate, control, monitor and review interactions with their data across all stages of the research.110
The International Ethical Guidelines also state that researchers should, through an “opt-out procedure,” inform persons whose data may be used in the context of research in the online environment of the purpose and context of the intended data uses, the privacy and security measures used to protect such data, and the limitations of the measures used and the privacy risks that may remain despite the implementation of safeguards.111 If a person objects to the use of his or her data for research purposes, the International Ethical Guidelines would forbid the researcher from using that data.112
In addition to the ethical principles set forth in the Declaration of Helsinki and the International Ethical Guidelines, a number of U.S. federal and state laws impose privacy- and security-related obligations on certain research studies or certain classes of researchers. For example, the Common Rule requires IRBs that review and approve research funded by a signatory agency to determine, when appropriate, that “adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data” exist.113 Similarly, the HIPAA Privacy Rule requires covered entities114 to adhere to certain use and disclosure requirements,115 individual rights requirements,116 and administrative requirements117 during the conduct of research. Under the HIPAA Privacy Rule, researchers working for covered entities must obtain prior written authorization from each research participant before using or disclosing the participant’s protected health information (PHI) unless the use or disclosure falls into one of four research-related exceptions to the authorization requirement.118 Moreover, the HIPAA Security Rule requires researchers working for covered entities to adhere to certain administrative,119 physical,120 and technical121 safeguards designed to ensure the confidentiality, integrity, and availability of electronic protected health information (ePHI) and to protect against reasonably anticipated threats or hazards to the security and integrity of ePHI.122 Finally, the HIPAA Breach Notification Rule requires researchers working for covered entities to provide certain notifications in the event of certain breaches of unsecured PHI.123
In light of the ethical and legal principles discussed above, mobile health researchers should implement reasonable privacy and security measures during the conduct of mobile health research. For example, some generally applicable privacy measures include reporting their study results without any individually identifying information, not permitting research results to be used for marketing and other commercial secondary uses without prior explicit consent from each research participant, and not using “click-through” or other non-explicit forms of consent. With regard to security, mobile health researchers should implement reasonable administrative, physical, and technical safeguards designed to protect the security of participant data, such as by safeguarding their physical equipment from unauthorized access, tampering, or theft; and encrypting research data or otherwise making data unintelligible to unauthorized users.
E. Heightened Obligations
The Common Rule recognizes several categories of participants whose vulnerabilities require careful assessment in the research context, including people with diminished capacity to make decisions about participating in research, such as children; those who may lack the autonomy to make decisions due to the institutional context in which the research would take place, such as prisoners or students; and pregnant women for whom decisions would affect both themselves and their fetus.124 In the context of unregulated mobile health research investigators are not legally bound to follow federal regulations and definitions of vulnerable populations, although many of the same populations should be regarded as potentially vulnerable to research-related harms, thereby obligating researchers to develop safeguards for their inclusion in health research.
Researchers also have greater ethical responsibilities when health research involves sensitive topics or participants with vulnerabilities that may or may not align with the populations identified in federal research regulations.Reference Coleman, Metcalf and Crawford125 Specifically, health research using mobile technologies that involves potentially sensitive or stigmatizing information, such as mental, sexual, or reproductive health information, warrants heightened attention to protect individuals’ privacy, confidentiality, and security, as noted in the International Ethical Guidelines.126 Further, mobile technology-mediated research poses a unique challenge in authenticating participants’ identities that does not exist when research is conducted face-to-face. Researchers establishing this authentication process must be particularly vigilant when it comes to assessing prospective participants’ age and capacity to consent to participate in health research.127
Additionally, unregulated health research may create or exacerbate the potential for other harms to individuals and groups. For example, groups that disproportionately rely upon mobile devices for access to the internet, such as those with a low-income, members of racial minorities, and rural residents, may be more vulnerable in the context of mobile health research.Reference Anderson and Sylvain128 Further, people with rare diseases and mental health conditions may be more likely to be identifiable through digital phenotyping, which involves quantification of granular information about individuals using active and passive data collected from mobile and wireless devices.Reference Montgomery, Chester, Kopp, Senders and Broekman129 Therefore, app developers and unregulated researchers should carefully assess whether any groups face vulnerability to research-related harms and, if so, ensure that using their information in unregulated mobile health research does not reinforce old forms of discrimination or health disparities, or generate new ones.Reference Reyes130
F. Independent Ethics Review
Independent oversight of biomedical and behavioral research, widely recognized as an international norm,131 provides fundamental protection for human research participants. Oversight bodies serve to assess the ethical acceptability of research, evaluate compliance with applicable laws and regulations, and guard against researchers’ biases.Reference Grady132 In the U.S., oversight by an IRB is required for research conducted or funded by the federal government, as well as research under the jurisdiction of the FDA. In general, IRBs are charged with prior review of research involving human subjects to ensure that risks are minimized and are reasonable relative to anticipated benefits, participants are selected equitably, informed consent is sought and documented as appropriate, and there are adequate provisions for monitoring participant safety and for protecting their privacy and the confidentiality of their data.133
Researchers whose studies are not subject to these regulations may voluntarily seek IRB review (e.g., from an independent IRBReference Forster134) to obtain ethical oversight as well as meet journal requirements to publish their results. However, researchers choosing to forgo IRB review would not be in violation of legal requirements.
There are several significant reasons why some form of independent oversight would be beneficial for much unregulated research.Reference Beskow135 First, many researchers are unable to objectively and reliably assess and monitor the ethical issues surrounding their own research. Second, whether or not research is technically subject to regulation, the same basic principles and requirements for the ethical conduct of research still apply.Reference Emanuel, Wendler and Grady136 Third, given the specific challenges and shortcomings in obtaining effective informed consent in unregulated health environments, protections beyond consent take on even greater importance.
A range of alternative approaches to independent oversight, both formal and informal, have been proposed. Examples include an oversight board established specifically for unregulated researchers; a forum through which researchers could get feedback and consultation from experts; and ethics training and formal certification for researchers as a replacement for independent oversight. Questions abound concerning any such approach and significant work would be needed to identify and develop effective models that are acceptable to all stakeholders, standardized, sustainable, and can be evaluated.
Strong arguments concerning the need for independent oversight notwithstanding, there are reasonable questions about the ability of traditional IRBs to serve in this role for unregulated mobile health research. Criticisms of traditional IRBs highlight time consuming and/or low-quality review processes, excess focus on consent forms, and lack of validated measures of IRB performance leading to unjustifiable variability in IRB procedures and decision-making.Reference Friesen, Redman and Caplan137 Critics also claim that there is little evidence concerning the actual protection provided to participants.Reference Emanuel138 Regarding review of studies involving mobile apps and devices, empirical research suggests IRB professionals may be unfamiliar with these novel technologies, uncertain about the risks involved, and unclear on how to become informed — all of which could lead to delays and variability in IRB review.Reference Nebeker139
A range of alternative approaches to independent oversight, both formal and informal, have been proposed. Examples include an oversight board established specifically for unregulated researchers; a forum through which researchers could get feedback and consultation from experts; and ethics training and formal certification for researchers as a replacement for independent oversight.140 Questions abound concerning any such approach and significant work would be needed to identify and develop effective models that are acceptable to all stakeholders, standardized, sustainable, and can be evaluated. The history of abuses in research with human subjects in the U.S. and around the world has amply demonstrated the limits of relying on researchers to self-regulate as a way to protect participants and their data.
G. Responsible Conduct and Transparency
All researchers, regulated or not, have ethical obligations for responsible conduct of research141 and transparency.Reference Nicholls, Shamoo and Resnik142 The obligation of all researchers to conduct their research responsibly fundamentally distinguishes scientific from non-scientific inquiry. This mandate includes appropriate study design, proper data gathering and analysis, and data sharing and publication practices. Other issues under responsible conduct of research include conflicts of interest, author credit on publications, intellectual property, data integrity, and plagiarism.Reference Martinson, Anderson and de Vries143
From the original Declaration of Helsink in 1964 to the modern open science movement,144 transparency has been recognized as essential to ensuring the reliability of scientific outputs. However, lack of transparency regarding matters such as research funding and secondary uses of data, seriously erode trust in research. Importantly, the ethical obligation for disclosure of relevant information about health research extends beyond participants (who traditionally receive disclosures in the informed consent process) to the research community and the public. Whereas a lack of transparency to participants goes to the validity of consent, the lack of transparency to the research community and the public goes to the legitimacy of the scientific inquiry and the validity of research findings. The ethics of transparency have been codified by groups ranging from the World Health Organization’s Code of Conduct for Responsible Research145 to the grassroots cybersecurity research movement, I Am the Cavalry’s Hippocratic Oath for Connected Medical Devices.Reference Woods, Coravos and Corman146
There are several unique barriers faced by unregulated health researchers as they attempt to uphold their ethical obligations for responsible conduct and transparency. First is the “black box” problem. It can be difficult even for experienced researchers to understand the engineering that underlies mobile health tools. Which data are collected, how they are stored, and with whom they are shared may be obscurely presented, if at all. When developing mobile health devices, researchers may not realize the “hackability” of their devices,Reference Newman147 thereby permitting the unethical exploitation of their devices. In addition, mobile health data may collect far more information than needed to accomplish the researcher’s goals; for example, recording an individual’s exact GPS coordinates when a less precise displacement vector would do.
H. Proposed Ethical Frameworks
Beyond the ethical principles discussed above, a range of other principles have been proposed to guide the use of digital health data in unregulated research. These include, but are not limited to, recommendations that digital health information be accurate;148 that experts (in experimental design, data analysis, research ethics) be accessible;Reference Grant, Wolf and Nebeker149 and that the most appropriate ethical frameworks/governance structures for any given project will vary depending on the characteristics of the researchers, participants, and research design.Reference Lynn150
Differences between traditional research and citizen/community/patient-directed studies have led some to question whether the traditional paradigm of ethical review (e.g., IRB/REC involvement) is appropriate in participant-led initiatives.151 Some have argued that IRB/REC involvement may “promote decisions specific to data ownership, data management, and informed consent that directly conflict with the aims of research that is explicitly participant-led.”152 In response, several more fluid and adaptable approaches have been put forth.
Some scholars153 have proposed a citizen science governance framework that exists along a continuum in which “people-related” choices (e.g., regarding project membership and privacy of members personal data) and “information-related” decisions (e.g., privacy of, access to, and ownership of data) are made using a more rigid top-down approach (e.g., platform developer) or more flexible, bottom-up (e.g., project managers) approach depending on the specific needs and goals of the project. In determining the most appropriate framework, some commentators recommend that studies make explicit the “full spectrum of meanings of ‘citizen science,’ the contexts in which it is used, and its demands with respect to participation, engagement, and governance.”Reference Woolley154
Other experts suggest that the specific ethics/governance expectations and obligations be, in part, determined by the researcher context; specifically, whether unregulated researchers are operating within state-recognized or state-supported institutions and/or are engaged in profit-making.155 When research occurs within such institutions or for-profit, then standard ethics review (identical obligations of oversight) would be appropriate.156 A risk-based approach can be used to divide all other types of projects (non-institutional and non-profit) into two categories. Studies in which the research involves more than minimal risk should require some form of ethics review, possibly equivalent to expedited review, or through open protocol crowd-sourcing ethics review. Studies involving no more than minimal risk would not require formal ethics review, but would still require oversight with respect to basic ethical principles and legal requirements.Reference Woods, Corvasos and Corman157 A range of ethical approaches also may be gleaned from international sources.Reference Lang, Knoppers, Zawati, Dove and Chen158 After reviewing these many sources of ethical frameworks, we have used fundamental ethical and policy considerations to guide our recommendations.
III. Ethical Issues and Policy Recommendations
A. Introduction
The opinions of policymakers, stakeholders, academics, and others on unregulated health research diverge widely. On the one hand, some experts advocate extending the Common Rule to all researchers, arguing that regardless of the funding source all research participants should be entitled to the same protections, such as a balancing of risks and benefits, informed consent, and confidentiality. Some of these experts take an all-or-nothing approach. If political considerations make it impossible to obtain comprehensive coverage under the Common Rule, they reject the idea of accepting lesser protections, such as voluntary ethics consultation for researchers and optional external ethics review, because they believe it erroneously assumes that partial protections are sufficient. Some view these measures as a “watered-down version of the Common Rule.”
On the other hand, many unregulated researchers and their advocates strongly object to any regulation or governmental involvement in unregulated health research, including mobile device-enabled research.159 They view regulation of this research as unnecessary and burdensome governmental meddling into valuable scientific inquiry. Some even oppose optional government consultation or educational assistance to unregulated researchers on the grounds that it is the first step to regulation.
After careful consideration, we decline to endorse either of these positions toward unregulated research. In the sections that follow, we make the case for a middle ground approach based on pragmatism. We recognize that such a position requires a deft balancing of all interests and that our position is susceptible to criticism from both sides of the issue. To address both sides, we begin with the argument that there is no need to have any new efforts directed at unregulated health research, including research using mobile devices and health apps.
In our view, the current laissez faire approach to unregulated health research in the U.S. is not in the best interests of participants, researchers, or the public. We begin by noting that most other countries regulate all biomedical research regardless of the funding source,160 and therefore the U.S. is an international outlier in this regard. Nevertheless, recent experience with the Common Rule amendment process (discussed in the following section) makes it highly unlikely that in the foreseeable future Congress will extend the Common Rule to all research. There might be some expansion of state research laws, but the likelihood and desirability of state legislation and enforcement in this area is unclear. In the current political atmosphere, we believe that sensible, reasonable, and demonstrably effective measures, though inferior to comprehensive coverage of the Common Rule, are still far superior to doing nothing. It also could be asserted that many of our recommendations to assist unregulated researchers should be available to regulated researchers as well. We do not quarrel with that view; we merely note that our task is to address unregulated research using mobile devices and not to address all of health research.161
We similarly reject the position of many unregulated researchers that an increased emphasis on research safeguards and ethical conduct is unnecessary. Although unregulated research using mobile devices rarely involves invasive or high-risk procedures, it still may cause a variety of harms.162 At a time when unregulated research is expanding, it is necessary and appropriate to consider a wide range of measures to protect the interests of research participants and the public. No researchers, regardless of their funding, training, or motivation should engage in conduct that creates unreasonable risks to research participants, and oversight is a key way of ensuring ethical grounding of all research with human participants.Reference Rasmussen163
Our recommendations utilize a combination of methods, including education, consultation, transparency, self-governance, and regulation. We support a risk-based approach to research ethics oversight whereby all no-risk or minimal-risk research would be exempt or subject to expedited ethics review. This principle, as applied to unregulated research, means that the level of risk would determine the degree to which traditional research ethics requirements apply.164 We believe that in the absence of expanded coverage of the federal research regulations the measures that follow will help protect participants in unregulated health research using mobile devices while still facilitating innovative methods of scientific discovery.
B. Federal Research Regulations
The National Research Act165 was enacted in 1974 in the aftermath of public disclosures and congressional hearings documenting the outrageous and unethical research practices involved in the Tuskegee Syphilis Study.Reference Jones and Brandt166 The Department of Health, Education, and Welfare (HEW) first published regulations for the protection of human subjects in 1974.167 The Department of Health and Human Services (HHS), the successor to HEW, led an inter-agency process that culminated in 1991 with publication of regulations for research conducted or funded by signatory federal departments and agencies.168 Because of their broad applicability, the Federal Policy for the Protection of Human Subjects became known as the “Common Rule.” The jurisdictional basis of the Common Rule was the federal government’s conduct or funding of the research. Separate regulations were promulgated by the Food and Drug Administration (FDA) in 1981, applicable to research conducted in anticipation of a submission to the FDA for approval of a drug or medical device.169
When it was originally adopted in 1991, the Common Rule’s coverage of federally-funded researchers was generally considered sufficiently comprehensive because the predominant model of research, especially biomedical research, involved centralized research at large institutions. These recipients of federal funding also generally agreed to abide by the Common Rule in all research conducted at their institutions, regardless of the funding source.170 The Institute of Medicine,Reference Federman, Hanns and Rodriguez171 the National Bioethics Advisory Commission,172 and other expert bodies have proposed that the federal research regulations should apply to all human subject research regardless of the funding source. The recent growth in unregulated research described in this article has added another dimension to this ongoing policy debate.
The lengthy and contentious rulemaking culminating with the recent revisions to the Common Rule,173 published in 2017 and effective in 2019,174 illustrates the difficulty in expanding the scope of the federal research regulations. In 2011, HHS, in coordination with the White House Office of Science and Technology Policy, published an Advanced Notice of Proposed Rulemaking, requesting public comments on how the existing federal research regulations might be modernized and improved.175 One specific area in which comment was sought was extending the Common Rule to all studies, regardless of the source of funding. In 2015, the Common Rule agencies issued a Notice of Proposed Rulemaking,176 which limited the proposed expansion of the Common Rule to all clinical trials or alternatively those clinical trials presenting greater than minimal risk, regardless of the funding.177 By 2017, the final rule issued by the Common Rule departments and agencies abandoned altogether the proposal to expand the coverage of the Common Rule.178
In light of this recent experience and the lack of political and public support for such a fundamental change, we have chosen not to focus our recommendations on expanding the coverage of the Common Rule to include all biomedical research regardless of the funding source.Reference Meyer179
C. State Research, Data Protection, and Genetic Testing Laws
1. state research laws
Recent state legislative activity in the areas of research regulation and consumer protectionReference Tovino and Tovino180 indicates a greater willingness of states to become involved with these issues, in part because of inaction by Congress. Because mobile research applications can collect data from participants who reside in different states, uniformity of laws (and uniformity of interpretation of such laws) is critical for implementation and compliance. Therefore, if state regulation is viewed as the best way to obtain comprehensive regulation of health research, the adoption of a model or uniform state law is preferable to wildly varying state enactments. Of the state research laws enacted thus far, we believe the Maryland law is the best.
a. Maryland
In 2002, Maryland enacted its state research law for “the purpose of requiring a person conducting human subject research to comply with federal regulations on the protection of human subjects.”181 Accordingly, Maryland regulates “all research using a human subject,” regardless of whether such research is federally funded,182 and prohibits “a person” from “conduct[ing] research using a human subject unless the person conducts the research in accordance with the federal regulations on the protection of human subjects (the Common Rule).”183
One reason the Maryland law is desirable in the context of mobile device-mediated health research is its unrestricted use of the word “person.” The Maryland law applies to all researchers, including traditional scientists, independent scientists, citizen scientists, and patient researchers, as well as any other person who conducts research.184 Other state research laws discussed below apply to a narrower class of researchers, such as researchers who are licensed physicians or researchers who conduct research in a licensed health care facility.
A second desirable feature of Maryland law is its definition of “federal regulations on the protection of human subjects.” The definition specifically references “Title 45, Part 46 of the Code of Federal Regulations [the Common Rule], and any subsequent revision of those regulations.”185 The Maryland law anticipates the possible revision of the Common Rule and expresses a clear desire for Maryland research to be conducted in accordance with the most current version of the Common Rule.
If the Maryland law were used as a model for other states, it would promote uniform requirements and protections with both the federal Common Rule and state research laws. As with all issues of federalism, however, the downside of uniformity and preventing a patchwork of state laws is that it prevents other states from adopting innovative approaches. Pioneering research laws implemented in state “laboratories of democracy”186 might identify improved ways to address emerging issues, such as health research with mobile devices.
b. Other State Research Laws
Six other states, Virginia, New York, California, Illinois, Wisconsin, and Florida, also have enacted research laws. None of these laws address the unique features of mobile device-mediated research. They also are not comprehensive or provide weaker protections for research participants than the Common Rule.
Of these other state laws, Virginia provides the most comprehensive coverage for non-federally funded human research,187 including detailed requirements for the formation of human research review committees,188 criteria for review committee approval of research,189 and mandatory provisions for informed-consent-to-research statements.190 Compliance with unique state laws would prove difficult for mobile device-mediated health researchers who collect data from study participants residing in various states.
The New York research law establishes a policy of protecting state residents against “pain, suffering or injury resulting from human research conducted without their knowledge or consent.”191 However, the New York law narrowly defines “human research” as investigations involving physical or psychological interventions.192 The New York law would thus leave unprotected participants of mobile device-mediated, solely information-gathering research studies.
The California Protection of Human Subjects in Medical Experimentation Act193 establishes a detailed “bill of rights”194 and a series of explicit informed consent requirements195 designed to benefit subjects of medical experiments,196 as well as damages for research conducted without consent.197 The law, however, only applies to “medical experiments” and would not protect participants of mobile device-mediated informational research studies.
The Illinois Act Concerning Certain Rights of Medical Patients applies only to physician-researchers who conduct research programs198 involving hospital inpatients or outpatients and therefore would not apply to most participants in health research using mobile devices because they are not hospital inpatients or outpatients.
The Wisconsin Patients’ Rights law199 only protects “patients,” defined as certain individuals with mental illness, developmental disabilities, alcoholism, or drug dependency who receive treatment for such conditions in certain licensed health care facilities.200
Finally, Florida’s Patient’s Bill of Rights and Responsibilities Act,201 only applies to patients of licensed health care providers and health care facilities.202
2. state data protection laws
In addition to state research laws, many states have data breach, data security, and data privacy laws that are potentially applicable to mobile device-mediated research.203 In particular, all fifty states and the District of Columbia have enacted data breach notification laws that require the notification of data subjects of certain informational breaches in certain contexts.204 In addition, thirty-six jurisdictions have enacted statutes designed to protect the security of certain data sets, and fifteen jurisdictions have enacted statutes designed to protect the privacy of certain data sets.205 In some states, these statutes already apply to mobile device-mediated researchers who conduct informational health research.206 In other states, minor amendments to the definitions of “covered entity,” “personal information,” and “doing business in the state” would be necessary before the statutes would apply to mobile device-mediated health research.207
Box 1 Recommendations for the States
1-1. States that do not currently regulate all non-federally funded research should consider enacting a comprehensive law (or amending existing laws) to regulate all research conducted in the state.
1-2. States considering such legislation should review the Maryland research law, which contains a broad definition of “person” performing research and expressly applies the most recent version of the Common Rule.
1-3. States should consider extending the application of data breach, data security, and data privacy statutes to all mobile device-mediated research.
1-4. States should consider extending the application of genetic testing laws to all research conducted in the state.
A concern about both state research and data protection laws is that unregulated researchers are unlikely to know that such laws even exist, and therefore public education programs should be part of any legislative strategies.
3. state genetic testing laws
Many states have laws regulating genetic testing that may be relevant to unregulated health research using mobile devices, even if the testing is performed by a DTC genetic testing company or other entity unaffiliated with the researchers. Among the most common types of provisions are those requiring informed consent,208 establishing the privacy and confidentiality of genetic information,209 and prescribing certain retention or disclosure practices.210
D. National Institutes of Health
The National Institutes of Health (NIH) is the world’s largest public funder of biomedical research, with a 2019 research budget of $39.2 billion.211 More than 80% of the research budget funds extramural research.212 Beyond its size and budget, there are additional reasons why NIH would be a logical entity to play a leading role in health research conducted by unregulated researchers using mobile devices. First, NIH currently has numerous programs promoting the development of novel and emerging research strategies, such as its Common Fund initiatives.213 Second, NIH has a variety of programs for scientific education and workforce development, including science education resources for students and educators.214 Third, NIH already has demonstrated an interest in mobile health215 and citizen science216 through ongoing programs, and as evidenced by funding this grant through the National Cancer Institute, National Human Genome Research Institute, Office of Science Policy and Office of Behavioral and Social Sciences Research in the Office of the Director.217
There are three main reasons why some individuals and groups might not view NIH as an appropriate entity to play a leading role in this area of research. First, NIH may be regarded as epitomizing the traditional research establishment to which many citizen scientists, DIY researchers, self-experimenters, and other unregulated researchers object. Second, NIH maintains a detailed system of compliance and oversight for its extensive grant portfolio, and the prospect of NIH — even symbolically — knocking on the door of every basement and garage laboratory would be most unwelcome. Third, NIH is not a source that independent health app developers would likely consult to obtain source data and guidance on developing apps used for health research.
The recommendations that follow propose that NIH expand its efforts to assist unregulated researchers, research participants, and health app developers. NIH’s first priority in this area should be to serve as a repository of information essential to all stakeholders in unregulated health research. We recommend that an advisory board of diverse stakeholders (e.g., citizen scientists, DIY researchers, patient-directed researchers, app developers) be appointed to assist NIH in its activities, thereby providing practical information and enhancing the credibility of NIH’s efforts. NIH should fund studies on unregulated health research to determine the most effective ways of encouraging voluntary adoption of best practices and developing open-source tools.
We believe these concerns can be addressed. We envision that the role of NIH would be limited to serving as an information clearinghouse, supporter of research infrastructure development, and convener working with a range of governmental and nongovernmental groups and individuals. NIH would not have any regulatory role nor would it be involved in the direct funding of unregulated research. NIH would maintain a low profile and a light touch in promoting quality in unregulated health research and in safeguarding the welfare of research participants. This limited role for NIH is in keeping with its extant legal authority. Although the goals for the conduct of regulated and unregulated research are aligned, alternative procedures are necessitated by the current legal provisions. It remains to be seen how effective alternative means would be when applied to unregulated researchers; nevertheless, measures adopted to aid unregulated researchers (e.g., training programs) could serve as a way to assess the efficacy of similar measures for regulated researchers.
The recommendations that follow propose that NIH expand its efforts to assist unregulated researchers, research participants, and health app developers. NIH’s first priority in this area should be to serve as a repository of information essential to all stakeholders in unregulated health research. We recommend that an advisory board of diverse stakeholders (e.g., citizen scientists, DIY researchers, patient-directed researchers, app developers) be appointed to assist NIH in its activities, thereby providing practical information and enhancing the credibility of NIH’s efforts. NIH should fund studies on unregulated health research to determine the most effective ways of encouraging voluntary adoption of best practices and developing open-source tools. In consultation with the Office for Human Research Protections (OHRP), NIH should work to create and disseminate educational tools about research protections. In consultation with OHRP, and with input from grantees, NIH should also study the feasibility of supporting cost-free, independent, external research review organizations to advise unregulated health researchers how to ensure that all their research is consistent with essential ethical principles. An alternative model with less direct involvement of NIH is for grantees to take the lead in information sharing, education programs, and consultation services for unregulated researchers.218
Finally, surveillance is a cornerstone of public health efforts to assess trends over time and then evaluate the impact of interventions.Reference Gostin and Wiley219 This concept, however, has not yet been applied to mobile health research because there is no specific reporting of mobile health research and no required adverse event reporting for unregulated researchers. Therefore, a surveillance program should be established to estimate the amount of mobile health research, including unregulated health research over time, as even those basic figures are not known. Surveillance also would include assessing and categorizing adverse events using rigorous standards for case definitions. Over time, these data can be used to improve how mobile research applications are developed and used in research, with the ultimate goal of reducing adverse events.
Box 2 Recommendations for the National Institutes of Health (NIH)
2-1. NIH should expand its support for unregulated health researchers and centralize responsibility for providing assistance. NIH may accomplish this by establishing a new Office of Unregulated Health Research, designating an existing Institute or Center to oversee initiatives on unregulated health research, funding grantees to provide assistance to unregulated researchers, or through other means.
2-2. NIH should appoint an advisory board of diverse stakeholders to assist the NIH official or entity in charge of unregulated health research.
2-3. Unregulated health researchers need accessible, consolidated, updated, and curated information about research laws and ethical considerations from a trusted source. NIH should provide technical and understandable information about mobile and wireless technologies for app developers, researchers, and research participants. Therefore, NIH should develop and maintain a website containing the following.
Information and FAQs identifying the laws applicable or inapplicable to health research, including the Common Rule, FDA, FTC, state research laws, and the HIPAA Privacy Rule;
Information about externally developed best practices and ethical principles for unregulated health research;
Directory of open source tools for health research apps, including sample consent documents, privacy protection measures, and security information; and
Directory of resources for technical assistance.
2-4. NIH should fund studies on unregulated mobile health research to determine the most effective ways of encouraging compliance with best practices, attaining and maintaining quality, and developing open-source tools.
2-5. NIH should, in consultation with OHRP, work with citizen science groups and other organizations of unregulated researchers to support educational programs for mobile health app developers, unregulated researchers, and participants, as well as to provide technical support.
2-6. NIH, in consultation with OHRP, should consider the feasibility of establishing or supporting cost-free, independent, research review organizations to advise unregulated researchers on identifying and resolving ethical challenges raised by their research.
2-7. NIH, in consultation with OHRP, the National Science Foundation (NSF), and other public and private entities should support the establishment of a surveillance system to monitor, categorize, and track the rate of health research using mobile devices. The surveillance system also should capture the incidence and nature of adverse events caused by health research using mobile devices in both regulated and unregulated research.
E. Food and Drug Administration (FDA)
The Food and Drug Administration (FDA) has jurisdiction over research with human participants in two main ways. First, clinical research conducted in contemplation of a submission to the FDA for approval of a drug or medical device is subject to detailed regulations similar to the Common Rule.220 Among other things, the regulations require informed consent,221 review by an IRB,222 and disclosure of investigators’ financial conflicts of interest.223 Second, the FDA has jurisdiction over medical devices that may be used in research, which potentially creates a pathway for the FDA to regulate research with human participants conducted by unregulated researchers. This section and the recommendations that follow are concerned with this second aspect of FDA jurisdiction.
The Food, Drug, and Cosmetic Act (FDCA), as amended,224 provides the FDA with a broad public health mandate that includes regulation of medical devices. Congress defines devices that are subject to FDA regulation as including any “instrument, apparatus, implement, machine, contrivance…” that is “[i]ntended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals” and components and accessories of such devices also are regulable as devices.225 Mobile health apps potentially fall within the scope of this definition. In recent years, this created concerns that the cost and burdens of FDA oversight might chill innovation in mobile medical applications and wellness products that could benefit consumers.Reference Sharkey226
To allay these concerns, the FDA issued guidance documents in 2013 and 2015 indicating its intent to regulate mobile apps only if they performed medical device functions that could pose a risk to patient safety if the mobile app failed to function as intended.227 In 2016, the 21st Century Cures Act228 formalized this policy by amending the FDCA’s definition of a device to remove five categories of software from the FDA’s jurisdiction.229 One of the exclusions relates to software for encouraging wellness or a healthy lifestyle, provided the software does not cross the line into “diagnosis, cure, mitigation, prevention, or treatment of a disease or condition.”230 This category, often referred to as general wellness software, includes such things as fitness trackers and medical calculators (e.g., for body mass index).
In 2019, the FDA issued final guidance that further clarifies the line between regulated and unregulated wellness software after 21st Century Cures.231 This is part of a larger, ongoing effort that the agency launched several years ago to implement a Digital Health Innovation Action Plan and create a Center of Excellence in Digital Health to improve its oversight of software.232 The FDA embraces a risk-based framework for regulating health information technology in concert with the Federal Communications Commission (FCC) and the Office of the National Coordinator for Health Information Technology (ONC), as directed by the Food and Drug Administration Safety and Innovation Act (FDASIA) of 2012,233 with the aim of limiting regulatory duplication or overstepping.234 Additionally, ONC, FDA, FTC, and the HHS Office for Civil Rights (OCR) have published an online, interactive decision tree to aid app developers in correctly identifying the federal laws that apply to their apps.235
We endorse the FDA’s focus on clarity and efficiency in digital health regulation, but there are unresolved issues relating to research that uses mobile health apps when — as often will be the case — such research is not regulated by the Common Rule. There is a risk that such research might encourage participants to use low-risk general wellness devices in new ways for which the devices are not safe and effective. This potential for repurposing and misuse exists because of the way the FDA determines whether a device is a low-risk general wellness device that is exempt from FDA oversight versus a medical device subject to FDA regulation. This determination does not necessarily reflect intrinsic properties of the device itself, in terms of the health characteristics that it is capable of measuring or the quality of the data it produces. General wellness devices sometimes produce data that closely resemble data from an FDA-regulated medical grade device.
For regulatory purposes, the distinction between a general wellness device and a medical device turns on the device’s intended use.236 The FDA’s algorithm for assessing the intended use of a device is set out in the agency’s regulation at 21 C.F.R. § 801.4. As a general matter, a device’s intended use refers to the objective intent of the “the persons legally responsible for the labeling”237 of the device, which in most instances is the device’s manufacturer. Section 804.1 allows the FDA to consider direct and/or circumstantial evidence of the manufacturer’s intent. Direct evidence would include claims the manufacturer or its representatives made about the device, such as in its labeling, advertising matter, and oral and written statements about the device.238 The allowed circumstantial evidence includes facts showing that the manufacturer knew that the device was being misused for purposes other than those for which it was labeled and advertised.239
In practice, however, it is unlikely that the FDA would hold a manufacturer responsible for other people’s misuse of its device, even if the manufacturer was aware of the misuse. As a leading treatise observes, “FDA has rarely attempted to classify a product as a drug or device in the absence of relevant representations by the manufacturer or distributor.”Reference Hutt, Merrill and Grossman240 In other words, the agency generally bases its decisions on the direct evidence: statements the manufacturer made in labeling, advertising, and marketing of the device. The FDA has never disclaimed its authority to establish a product’s intended use based on circumstantial evidence of a known misuse, but it is rare for the FDA to assert that authority,241 and even rarer for the FDA to prevail in court when it does.242 The recent FDA guidance on general wellness devices treats manufacturers’ claims about a device as the main source of evidence the agency will use to distinguish (unregulated) general wellness devices from (regulated) medical devices.243
A device that is lawfully on the market, but which is being used in research in a novel way that goes beyond its intended uses, potentially becomes an investigational device that requires an investigational device exemption (IDE).244 The IDE regulations aim to protect study participants when they are exposed to risk from devices being used in ways that the FDA has not established are safe and effective, and to ensure that the devices produce valid data. The IDE regulations apply to research sponsored by device manufacturers trying to generate data for submission as part of an FDA pre-market review, but the agency emphasizes that its IDE requirements also may apply in other research settings. There are many unresolved questions about whether and how these regulations would apply to unregulated health research using mobile devices.
Box 3 Recommendations for the Food and Drug Administration (FDA)
3-1. The FDA should continue its interagency collaborative efforts to reduce regulatory duplication and identify and assess areas unaddressed by current regulations. One area for immediate interagency consideration is how best to ensure transparency in validation of mobile health app algorithms, in situations where the FDA has jurisdiction to regulate such devices.
3-2. The FDA should increase its engagement with the health app developer community to raise awareness of its guidance documents, including those issued in September 2019.
3-3. The FDA should require developers of mobile health apps subject to its jurisdiction to make transparent disclosures regarding the intended use (including research) and technical capacities of their apps, especially mobile medical apps.
3-4. The FDA’s guidance documents do not address how its regulations apply to citizen-led research using data from mobile health apps. In particular, the FDA should clarify the following: (1) when such research may require an IDE; (2) what forms of research using data from mobile health apps constitute “significant risk” research under the IDE regulations; (3) how the concept of a “sponsor-investigator study” applies to nontraditional and citizen-led research; (4) what forms of communication about citizen science projects could subject organizers to charges of unlawful promotion of unapproved uses of a device; and (5) what constitutional constraints limit the FDA’s power to regulate nontraditional, citizen-led research efforts.
The FDA gains its authority to regulate research as an incident of its authority to regulate medical products such as drugs and devices. This implies that the FDA’s research regulatory mandate is narrower in scope than the Common Rule. Part 812 allows the FDA to regulate studies of devices (“clinical investigations of devices to determine safety and effectiveness”)245 but not studies that merely use devices as tools to explore basic scientific or medical questions. The agency’s own training materials state that no IDE is required for “basic physiological research” that is “investigating a physiological principle” with “no intent to develop the device for marketing,” if the investigation is “only using the device to address the research question.”Reference Henley246
Although the FDA generally does not regulate “investigations to expand medical knowledge or conduct fundamental research,”247 there are a few exceptions that allow the FDA to regulate such research. One exception is for broad scientific studies that incorporate a device study. “If the expansion of medical knowledge or the conduct of fundamental research involves an investigation to determine the safety or effectiveness of a device, an IDE will be required.”248 A second exception allows the FDA to require an IDE if a broad scientific study uses a device in novel ways that pose “significant risk” for the research subjects.249 In some contexts, the FDA has taken the position that research poses significant risk if patient-specific results are returned to participants. The FDA has not clarified how these principles apply to citizen science projects. Such studies often are observational rather than interventional in nature, so the risk they pose may be minimal. Moreover, it is hard to characterize research as “returning” patient-specific results to participants, when the participants collected the data and had possession of it in the first place.
Another regulatory risk involves unlawful promotion of unapproved uses of legally marketed devices.250 The IDE regulations prevent research sponsors, investigators, and persons acting on their behalf from communications that promote an investigational device that has not been cleared or approved251 or that represent the device as safe and effective for the purposes for which it is being studied.252 There is a risk that the FDA might deem a citizen science project to be promoting unapproved uses of general wellness devices. This could chill constitutionally protected scientific speech unless the boundaries of the FDA’s efforts to regulate such “promotion” are carefully drawn.
Many citizen science and other nontraditional research projects are “sponsor-investigator” studies in which the sponsor and the investigator are the same person. Sponsor-investigator studies are believed to pose special risks for human research participants, because they lack the inherent checks and balances of having a separate sponsor and investigator keeping an eye on one another’s activities. In 2005, an FDA official noted a high incidence of participant protection problems in such studies.253 Some of the FDA’s training materials suggest that the FDA can require an IDE for sponsor-investigator studies, regardless of whether the study sponsor aims to submit data to the FDA, to develop a new device for marketing, or to expand the intended uses of a legally marketed device.254 This position has been questioned,Reference Evans255 but the agency has not reversed it, and it adds to the regulatory uncertainty around citizen science projects.
F. Federal Consumer Protections through the FTC and CPSC
Federal agencies tasked with ensuring consumer protection with regard to commercial products and services, such as the Federal Trade Commission (FTC) and the Consumer Product Safety Commission (CPSC), are well-positioned to regulate mobile platform-based health research because they can take more comprehensive approaches to regulation.Reference Terry256 As mentioned above, the FDA’s jurisdiction does not extend to some mobile health apps’ software functions and uses, such as fitness trackers and medical calculators, and therefore regulatory responsibility could fall to consumer protection agencies.
The FTC is responsible for preventing unfair competition and unfair or deceptive acts or practices by entities engaged in or affecting commerce. It can seek monetary redress for consumer injuries, make legislative recommendations, and prepare reports. The FTC’s preventative mission positions it to use self-regulation enforcement mechanisms; this means that the FTC does not need to wait until consumers are harmed before it can act. By using selective enforcement within an industry the FTC can protect mobile technology-mediated health research participants, including their health privacy, through regulation of unfair trade practices.Reference Hoofnagle257
The FTC could be particularly useful in regulating the adequacy of consumer technology companies’ and app developers’ privacy policies and practices, transparency, and fairness practices in the research they undertake or facilitate. The FTC has brought enforcement actions against flagrant offenders of consumer protection in the health space since 2011, with a particular focus on commercial entities that have claimed to identify or cure health conditions such as acne, skin cancer, and vision problems.Reference Wagner258 Its enforcement actions have focused on truth-in-advertising, substantiation requirements to support product claims, and privacy and security breaches and insufficiencies.259 Further, the FTC co-produced a web-based interactive tool with HHS, ONC, OCR, and FDA, housed on the FTC website, to guide app developers about the federal laws that apply to the development and implementation of mobile health apps.260 These efforts indicate that the FTC is familiar with and monitoring mobile health, which could include mobile health research, for instance in surveilling the fairness of algorithms used and generated in unregulated mobile health research.
The CPSC is a federal agency authorized to protect consumers through surveillance functions and enforcement. In the digital arena, the CPSC issued a report to guide consumer safety and protection in relation to digitally connected devices entitled “A Framework for Safety for the Internet of Things.”261 In this framework the CPSC focuses on the potential for these devices to result in physical harms, illness, and death of consumers. Although narrow in scope, this framework illustrates that the CPSC sees the Internet of Things, within which mobile-platform enabled health research is a component, as within its jurisdiction. Furthermore, recognition that digital connectivity can result in psychological, emotional, and social harms that can be deeply injurious suggests that unregulated mobile health research could be subject to the CPSC’s regulation.
Box 4 Recommendations for the Federal Trade Commission (FTC) and the Consumer Product Safety Commission (CPSC)
4-1. The FTC should increase its efforts to encourage self-regulation of unregulated mobile health researchers by providing guidance and educational resources to app developers, unregulated researchers, and participants in unregulated mobile health research through, among other things, best practice guidelines and web-based, interactive educational tools.
4-2. The FTC should promote privacy, transparency, and fairness in unregulated mobile health research using preventative and remedial approaches, such as the following.
The FTC should increase targeted enforcement actions against developers of unregulated mobile health research platforms who engage in deceptive or unfair trade practices (e.g., making false or misleading statements, failing to provide adequate privacy or security for mobile Internet-connected devices) and seek monetary redress and other appropriate relief on behalf of injured consumers.
The FTC should develop and provide multi-media educational materials for consumers about the kinds of harms and complaints being monitored, and publicize bad actors in the unregulated mobile health research sector through consumer advisories.
4-3. The CPSC should increase surveillance and monitoring of research software, applications, and systems enabled through mobile, internet-connected devices by establishing a consumer hotline or website for reporting safety concerns, such as data breaches, and it should assess monetary penalties against researchers and developers who violate consumer product safety regulations pertaining to internet-connected mobile devices.
The CPSC’s jurisdiction may extend to circumstances and conditions not covered by the FDA, but the CPSC has limited regulatory tools. Thus, unless its regulations and enforcement powers are strengthened, merely having jurisdiction does not necessarily translate into the ability to exercise meaningful regulatory oversight.
G. Consumer Technology Companies and App Developers
Consumer technology companies fall along a spectrum of size and power, but taken together represent the dominant players in unregulated mobile health data collection. These companies are familiar with regulation through their interactions with Congress, the FCC, the FTC, and other government agencies, but the health policy issues represent a relatively novel space for most companies.
Consumer technology companies may be divided into two categories — platforms and startups — each of which has two sub-categories. We break down platforms into app stores and handset manufacturers, and startups into those making research apps and wearables. This division recognizes that companies that have achieved market dominance have significantly more economic power to create actual change than early stage startups, as well as more exposure to regulation via traditional channels over time. Startups also have radically different resource levels and incentives compared to platforms. In this division, we also note that the primary regulators of startups may well be the platforms that broker access to customers for the startup.
Apple is perhaps the clearest example of a consumer platform as regulator, recently acting to protect consumer privacy with a unilateral technical change around email login.Reference Glaser262 Within our topic area, Apple already encodes visual informed consent (i.e., an interface that requires app users to slow down and reflect on key issues through the use of icons, videos, and other methods)Reference Wilbanks263 into ResearchKit developer documentation and requires ResearchKit apps to have IRB approval regardless of whether the Common Rule applies.Reference Duhaime-Ross264
A wide variety of consumer technology platforms exist. Microsoft’s Windows is a platform, as is Amazon. For our purposes, we will focus on a very small subset of consumer technology companies that are most relevant to unregulated mobile health research: app stores as platforms, phone+wearables and their related mobile applications as platforms (i.e., from Apple or Google), research apps from startups, and “wearable” devices from startups. This focus excludes certain cases, but allows us to look at the vast majority of interactions with technology companies driving mobile research.
Platforms at their most basic are “digital infrastructures that enable two or more groups to interact.”Reference Srnicek265 The race to create a platform that enables many groups to interact defines much of the contemporary consumer technology space; massive amounts of capital pour into platforms to claim the network effect through subsidized products.Reference Parker and Van Alstyne266 The vast majority of consumer technology companies involved in unregulated health research aspire to platform status. Thus, for our purposes, what distinguishes a platform from a startup is simply the question of market adoption. Has the company achieved enough power to serve a soft-power regulatory function, or is the company likely to make choices in hopes of achieving that power while being regulated in turn by larger platforms? This type of ontology is necessary to delineate apps that are truly about self-tracking or “n of we” such as those seen in quantified self267 — primarily observational, with little aspiration to scale to thousands or millions or billions of users — from those funded by venture capital aspiring to monetize research data.
The app store duopoly puts Apple and Google in a de facto regulatory position, a situation that is not likely to change in the near future. Their differing review regimes represent forms of governanceReference Greene and Shilton268 that, for example, lead to a difference in the ability of developers to implement spyware or to submit “copycat” applicationsReference Rajasegaran, Liu and White269 ranging from innocuous clones to apps that actively install malware and spyware. Android users are also shown to be vulnerable to “grayware” — “potentially harmful apps” falling somewhere between utility and exploitation, typically just outside the illegal space.Reference Andow270 Our policy recommendations for these two companies therefore leverage this “soft power” to regulate the vast world of applications used in mobile health research.
Hardware manufacturers is a broad category. It includes phone handsets, although phones are often quite secure hardware platforms compared with other types of devices, such as the booming and varied field of “wearable health trackers.” Hardware companies must balance consumer-facing priorities and ease of pairing with an app on a mobile device with security features. New features that would significantly improve privacy and security — for example, encryption of all data “at rest” (on the device itself) typically drains battery performance,Reference Wander271 and thus is often left off devices during the design phase. Google’s choice not to mandate encryption on its Android phones by default, for example, has led to a gap with Apple phones in terms of encryption, with only around 10% of Android phones encrypted at rest versus nearly 95% of iPhones.Reference Afonin272 Google has responded to this gap, with Android Q poised to require forms of encryption across its ecosystem.Reference Porter273 These issues move beyond encryption and cover issues such as how devices are identified over time, how they integrate into other systems, and what data are gathered as part of quality control, each of which expose the user and the app to potential security attacks.Reference Cyr274
Complicating factors further is the widespread redistribution of wearable hardware data from the cloud systems of the hardware companies. Some hardware companies like Fitbit encourage users to access, share, and redistribute their own data, without discussions of how that access and redistribution can re-identify users via the mosaic effect.Reference Kugler and Strahilevitz275 This leads to outcomes such as Strava’s open data portal revealing the location of secret military bases via exercise patterns — with those devices now being banned for deployed troops.Reference Copp276 These data are also often widely re-sold or shared with third parties such as insurance plans,Reference Wischhover277 where they can represent emergent attack vectors for re-identification over time Reference Na278 (early research indicates only six days of full step counts are sufficient to re-identify an individual out of 100,000,000 participants).Reference Foschini279
H. Organizations of Unregulated Researchers
As previously mentioned, unregulated researchers include citizen scientists, DIY researchers, self-experimenters, and patient-based research networks that promote research among their members. Terms such as citizen scientist and DIY researcher originally signified that researchers were amateurs or lay-people conducting research without scientific training and outside of traditional research settings. Today, however, they also include individuals with scientific backgrounds working outside of traditional research settings, not-for-profit patient and research organizations, and corporate entities.284 Unregulated researchers may work independently, out of their homes or in community laboratories, or they may work in collaboration with more traditional regulated researchers.285 Some unregulated researchers, such as self-experimenters and crowd-sourcing data suppliers, may passively or actively participate in unregulated research efforts,Reference Bonney, Irwin and Wright286 giving existing mobile-platform data about themselves or collecting or generating new data via a mobile-platform to unregulated researchers. Previous research has shown that unregulated researchers are a heterogeneous group with varying degrees of relationship with regulated researchers and regulators.Reference Prainsack and Prainsack287
Although entities covered by the Common Rule have developed fairly uniform conceptions of the privacy protections and risks of participation in research with human participants, unregulated researchers and unregulated research organizations may consider the elements of traditional research ethics differently than regulated researchers.288 For instance, some unregulated researchers may value speed or openness of data collection and management to answer research questions over privacy of individual participants or eschew research funding so long as research questions can be answered efficiently.289 While they may lack a common vocabulary or set of values with regard to best practices for mobile health research, some shared features of citizen science organizations are their reliance on information communication technologies to collect, supply, and analyze data, crowdsourcing research participation, and “grassroots” strategies to fund research.290
Box 5 Recommendations for Consumer Technology Companies and App Developers
5-1. Google should join Apple in requiring a signed informed consent document for any mobile health research applications emerging from the use of ResearchStack.
5-2. Apple and Google should require developers to upload IRB approval letters as PDFs, and make those documents available in-line to consumers contemplating installing a mobile research app. This disclosure requirement is compatible with both traditional institutional review and with unregulated research where there is more than minimal risk.Reference Vayena and Tasioulas280
5-3. Apple and Google should implement and enforce a “floor” for privacy policies and terms of use. For example, such a floor could include provisions that no data may be transferred to third parties without specific consent for each use.281
5-4. Developers of research apps should leverage the existing, community-standard toolkits, such as ResearchKit and ResearchStack, each of which contains informed consent workflows and developer tools. These apps should (1) accommodate independent review when required by the app store platforms; (2) allow for isolation of malicious code elements; (3) publish a “software bill of materials” for any code integrated from a repository such as GitHub; and (4) publish a privacy disclosure notice.Reference Ciochetti and Kelley282
5-5. Makers of wearable devices should implement encryption both for data at rest and in transit. We further encourage federal and state investment in fundamental encryption research and development to support encryption on wearables that is easier to include for developers without overly damaging battery performance.
5-6. Security also should be implemented once the data have left the wearable and moved to the consumer’s phone (or directly to the servers of the wearable company), and therefore we recommend that manufacturers of wearables adhere to basic cybersecurity practices.Reference Anaya, Tahir, Tahir and McDonald-Maier283
Some organizations of unregulated researchers (e.g., PatientsLikeMe, Quantified Self, and the Citizen Science Association) are well positioned to promote best practices for mobile health-based research through education. For example, in 2011, DIYBio published a Draft Code of Ethics, including provisions regarding open access, transparency, education, and safety.291 These organizations also may serve as liaisons between the NIH, regulatory bodies, app developers, research participants, and individual unregulated researchers.
Box 6 Recommendations for Organizations of Unregulated Researchers
6-1. Organizations of researchers conducting studies in unregulated environments, such as community organizations, member associations, and patient research networks, should adopt guidance and/or standards for their members, including on the following issues:
Guidance on how best to transparently communicate the goals, risks, benefits, and data handling procedures of their research prior to enrolling a participant.
Guidance on privacy policies and terms of service for mobile device-based research.
Guidance on the privacy policies and terms of service of third party developed devices or apps.
Our recommendations propose that organizations of unregulated researchers should assume a greater role in educating and guiding their members about research.
IV. Conclusion
The general legal and ethical provisions for research with human participants in most of the world are easy to summarize. All researchers are legally required and ethically compelled to adhere to established rules and norms of research ethics. Thus, for example, researchers must minimize risks and maximize benefits to participants, participants must provide knowing and voluntary informed consent, and a research ethics committee must approve the research protocol. Things are more complicated in the United States, where federal research regulations apply to most, but not all, research. In the handful of states that have their own research laws, almost all of them are limited in scope, protections, or both. Thus, there is a discrete and growing category of “unregulated” research.
Recent technological and societal developments in the U.S. have increased the amount of unregulated research performed, although there are no estimates available on the amount of unregulated research or number of unregulated researchers. The widely assumed increase in unregulated research is attributable to, among other things, development of research apps for smartphones and other mobile devices, availability of direct-to-consumer genetic and other biomedical testing, formation of social media groups interested in health research, and growth of big data analytics for compilation and analysis of diverse data sets. The societal concern is that some unregulated research, mostly conducted by citizen scientists and others without formal training in scientific research, may be of low quality and suffer from a lack of ethics review, fail to adhere to generally recognized ethical norms for the treatment of research participants, and result in findings that expose research participants or larger groups of individuals (e.g., those with certain health conditions) to a range of harms from privacy violations to psychological and physical injury.
Extending federal research regulations to cover all research with human participants would be the easiest and most effective way to address the problem, but it is not viable politically and therefore it would be pointless to recommend. We also reject the view that the current laissez faire approach to unregulated research should be retained. Our preferred, middle ground option, is a suite of measures including education, consultation, transparency, self-governance, and regulation to ensure the quality of unregulated research and inform researchers and potential participants about key issues in research ethics. Our recommendations are directed to state governments, federal agencies, technology companies, app developers, and organizations of unregulated researchers.
The ethical bases of our recommendations are widely respected research ethics pronouncements in the U.S. (e.g., Belmont Report) and internationally (e.g., Nuremberg Code, Declaration of Helsinki). The unifying characteristic of our recommendations is pragmatism. We believe that our recommendations are realistic, feasible, and likely to produce positive results. They are also designed to be freestanding. Thus, even if all of our recommendations are not adopted, the adopted ones will make a valuable contribution to the conduct of unregulated health research using mobile devices. At the same time, implementation will require coordination and oversight to ensure that the multiple entities giving force to the recommendations do not establish conflicting regimes.
Acknowledgments
The authors are indebted to the following members of the grant’s working group, many of whom have written separate articles for this symposium or helped improve this article with their edits and comments: Kate Black, Shawneequa Callier, Ellen Wright Clayton, Ted Dove, Barbara Evans, Dov Fox, Malia Fullerton, Cindy Geoghegan, Aaron Goldenberg, Hank Greely, Christi Guerrini, Steve Hershman, Katie Huber, Marc Hurlbert, Eric Juengst, Bartha Maria Knoppers, Mary Majumder, Gary Marchant, Amy McGuire, Michelle Meyer, Camille Nebeker, Sally Okun, Pearl O’Rourke, Kevin Patrick, Patty Spears, Nic Terry, Sharon Terry, Jen Wagner, Susan Wolf, Joon-Ho Yu, and Ma’n Zawati.