










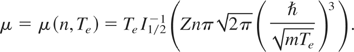
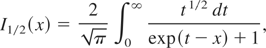
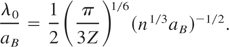















Crossref Citations
This article has been cited by the following publications. This list is generated based on data provided by Crossref.
Anisimov, S. I.
Zhakhovskiĭ, V. V.
Inogamov, N. A.
Nishihara, K.
Petrov, Yu. V.
and
Khokhlov, V. A.
2006.
Ablated matter expansion and crater formation under the action of ultrashort laser pulse.
Journal of Experimental and Theoretical Physics,
Vol. 103,
Issue. 2,
p.
183.
Canova, F.
Flacco, A.
Canova, L.
Clady, R.
Chambaret, J.-P.
PlÉ, F.
Pittman, M.
Planchon, T.A.
Silva, M.
Benocci, R.
Lucchini, G.
Batani, D.
Lavergne, E.
Dovillaire, G.
and
Levecq, X.
2007.
Efficient aberrations pre-compensation and wavefront correction with a deformable mirror in the middle of a petawatt-class CPA laser system.
Laser and Particle Beams,
Vol. 25,
Issue. 4,
p.
649.
Lin, Zhibin
and
Zhigilei, Leonid V.
2007.
Temperature dependences of the electron–phonon coupling, electron heat capacity and thermal conductivity in Ni under femtosecond laser irradiation.
Applied Surface Science,
Vol. 253,
Issue. 15,
p.
6295.
Anisimov, S. I.
Inogamov, N. A.
Petrov, Yu. V.
Zhakhovskii, V. V.
and
Nishihara, K.
2007.
Laser Ablation and its Applications.
Vol. 129,
Issue. ,
p.
1.
Veysman, M E
Agranat, M B
Andreev, N E
Ashitkov, S I
Fortov, V E
Khishchenko, K V
Kostenko, O F
Levashov, P R
Ovchinnikov, A V
and
Sitnikov, D S
2008.
Femtosecond optical diagnostics and hydrodynamic simulation of Ag plasma created by laser irradiation of a solid target.
Journal of Physics B: Atomic, Molecular and Optical Physics,
Vol. 41,
Issue. 12,
p.
125704.
Lee, Jae Bin
Kang, Kwangu
and
Lee, Seong Hyuk
2011.
Comparison of Theoretical Models of Electron-Phonon Coupling in Thin Gold Films Irradiated by Femtosecond Pulse Lasers.
MATERIALS TRANSACTIONS,
Vol. 52,
Issue. 3,
p.
547.
Inogamov, N. A.
Faenov, A. Ya.
Zhakhovsky, V. V.
Pikuz, T. A.
Skobelev, I. Yu.
Petrov, Yu. V.
Khokhlov, V. A.
Shepelev, V. V.
Anisimov, S. I.
Fortov, V. E.
Fukuda, Y.
Kando, M.
Kawachi, T.
Nagasono, M.
Ohashi, H.
Yabashi, M.
Tono, K.
Senda, Y.
Togashi, T.
and
Ishikawa, T.
2011.
Two‐Temperature Warm Dense Matter Produced by Ultrashort Extreme Vacuum Ultraviolet‐Free Electron Laser (EUV‐FEL) Pulse.
Contributions to Plasma Physics,
Vol. 51,
Issue. 5,
p.
419.
Inogamov, N. A.
Zhakhovsky, V. V.
Faenov, A. Ya.
Fortov, V. E.
Kato, Y.
Shepelev, V. V.
Fukuda, Y.
Kishimoto, M.
Nagasono, M.
Ohashi, H.
Senda, Y.
Yabashi, M.
Tono, K.
Tanaka, M.
Skobelev, I. Yu.
Pikuz, T. A.
Anisimov, S. I.
Petrov, Yu. V.
Ishino, M.
Ishikawa, T.
Togashi, T.
Nishikino, M.
Khokhlov, V. A.
Kando, M.
and
Kawachi, T.
2011.
Ablation of insulators under the action of short pulses of X-ray plasma lasers and free-electron lasers.
Journal of Optical Technology,
Vol. 78,
Issue. 8,
p.
473.
Inogamov, Nail A.
Zhakhovsky, Vasily V.
Petrov, Yurii V.
Khokhlov, Viktor A.
Ashitkov, Sergey I.
Migdal, Kirill P.
Ilnitsky, Denis K.
Emirov, Yusuf N.
Khishchenko, Konstantin V.
Komarov, Pavel S.
Shepelev, Vadim V.
Agranat, Mikhail B.
Anisimov, Sergey I.
Oleynik, Ivan I.
Fortov, Vladimir E.
Veiko, Vadim P.
and
Vartanyan, Tigran A.
2013.
Ultrashort laser-matter interaction at moderate intensities: two-temperature relaxation, foaming of stretched melt, and freezing of evolving nanostructures.
Vol. 9065,
Issue. ,
p.
906502.
Inogamov, N. A.
Zhakhovsky, V. V.
Petrov, Yu. V.
Khokhlov, V.A.
Ashitkov, S. I.
Khishchenko, K. V.
Migdal, K. P.
Ilnitsky, D. K.
Emirov, Yu. N.
Komarov, P. S.
Shepelev, V. V.
Miller, C.W.
Oleynik, I.I.
Agranat, M.B.
Andriyash, A.V.
Anisimov, S.I.
and
Fortov, V.E.
2013.
Electron‐Ion Relaxation, Phase Transitions, and Surface Nano‐Structuring Produced by Ultrashort Laser Pulses in Metals.
Contributions to Plasma Physics,
Vol. 53,
Issue. 10,
p.
796.
Inogamov, N. A.
Petrov, Yu. V.
Khokhlov, V. A.
Anisimov, S. I.
Zhakhovskiĭ, V. V.
Ashitkov, S. I.
Komarov, P. S.
Agranat, M. B.
Fortov, V. E.
Migdal, K. P.
Il’nitskiĭ, D. K.
and
Émirov, Yu. N.
2014.
The effect of an ultrashort laser pulse on metals: Two-temperature relaxation, foaming of the melt, and freezing of the disintegrating nanofoam.
Journal of Optical Technology,
Vol. 81,
Issue. 5,
p.
233.
Minakov, D. V.
and
Levashov, P. R.
2015.
Melting curves of metals with excited electrons in the quasiharmonic approximation.
Physical Review B,
Vol. 92,
Issue. 22,
Khokhlov, V A
Petrov, Yu V
Inogamov, N A
Migdal, K P
Winter, J
Aichele, C
Rapp, S
and
Huber, H P
2019.
Dynamics of supported ultrathin molybdenum films driven by strong short laser impact.
Journal of Physics: Conference Series,
Vol. 1147,
Issue. ,
p.
012066.
Coudert, Stéphane
Dilhaire, Stefan
Lalanne, Philippe
and
Duchateau, Guillaume
2022.
Unified theoretical description of out-of-equilibrium electron intraband dynamics in gold induced by femtosecond laser pulses.
Physical Review B,
Vol. 106,
Issue. 13,
Borah, Rituraj
Kumar, Ashish
Samantaray, Millen
Desai, Anusha
and
Tseng, Fan-Gang
2023.
Photothermal Heating of Au Nanorods and Nanospheres: Temperature Characteristics and Strength of Convective Forces.
Plasmonics,
Vol. 18,
Issue. 4,
p.
1449.
Winter, Jan
Redka, David
Minár, Ján
Schmidt, Michael
and
Huber, Heinz P.
2023.
Resolving transient temperature and density during ultrafast laser ablation of aluminum.
Applied Physics A,
Vol. 129,
Issue. 9,
Inogamov, N. A
Khokhlov, V. A
Romashevskiy, S. A
Petrov, Yu. V
Ovchinnikov, M. A
and
Ashitkov, S. I
2024.
SIL'NOE VOZBUZhDENIE ELEKTRONNOY PODSISTEMY ZOLOTA UL'TRAKOROTKIM LAZERNYM IMPUL'SOM I PROTsESSY RELAKSATsII OKOLO TEMPERATURY PLAVLENIYa.
Журнал экспериментальной и теоретической физики,
Vol. 165,
Issue. 2,
p.
165.
Feng, Jiecai
Wang, Junzhe
Liu, Hongfei
Sun, Yanning
Fu, Xuewen
Ji, Shaozheng
Liao, Yang
and
Tian, Yingzhong
2024.
A Review of an Investigation of the Ultrafast Laser Processing of Brittle and Hard Materials.
Materials,
Vol. 17,
Issue. 15,
p.
3657.