Significance outcomes
∙ Pre-treatment with sodium benzoate (SB), a prototype d-amino acid oxidase inhibitor, attenuated pre-pulse inhibition deficits and hyperlocomotion in mice after a single administration of phencyclidine.
∙ However, a single administration of SB did not affect d-serine levels in the blood and brain.
Limitations of the study
∙ In this study, we did not measure d-serine levels in the cerebellum where d-amino acid oxidase is rich.
∙ The effects of SB in other models of schizophrenia should be examined.
∙ The effects of chronic administration of SB on levels of amino acids in the brain should be examined.
Introduction
Multiple lines of evidence suggest that dysfunctional glutamatergic neurotransmission via N-methyl-d-aspartate (NMDA) receptors is involved in the pathophysiology of schizophrenia (Reference Javitt and Zukin1–Reference Hashimoto9). The NMDA receptor antagonists, phencyclidine (PCP), and ketamine induce schizophrenia-like symptoms, including positive and negative symptoms, and cognitive impairment in healthy individuals (Reference Javitt and Zukin1,Reference Krystal, Karper and Seibyl10–Reference Anand, Charney and Oren12). This resulted in the frequent use of PCP to generate animal models of schizophrenia (Reference Jentsch and Roth13–Reference Shirai, Fujita and Hashimoto22).
Accumulating evidence suggests that disturbed NMDA receptor neurotransmission, due to decreased d-serine levels, may be a causative factor in the pathophysiology of schizophrenia (Reference Hashimoto, Shimizu and Iyo6,Reference Hashimoto7,Reference Coyle and Tsai23–Reference Labrie, Wong and Roder25). These findings include, first, lower levels of d-serine in the blood, cerebrospinal fluid, and post-mortem brain tissue from patients with schizophrenia relative to normal controls (Reference Hashimoto, Fukushima and Shimizu26–Reference Calcia, Madeira and Alheira30). Second, treatment with d-serine reduces positive, negative, and cognitive symptoms in patients with schizophrenia (Reference Tsai, Yang, Chung, Lange and Coyle31–Reference Ermilov, Gelfin and Levin35). In addition, a recent meta-analysis supports the finding that d-serine is effective in the treatment of schizophrenia (Reference Tsai and Lin36). Third, mRNA expression and the activity of d-amino acid oxidase (DAAO), the enzyme that metabolises d-serine, is increased in post-mortem brain samples from patients with schizophrenia (Reference Verrall, Walker and Rawlings37,Reference Madeira, Freitas, Vargas-Lopes, Wolosker and Panizzutti38). Fourth, the G72 gene, located at chromosome 13q, shows significant association with schizophrenia (Reference Chumakov, Blumenfeld and Guerassimenko39,Reference Kvajo, Dhilla, Swor, Karayiorgou and Gogos40). This gene has been designated to code a DAAO activator, as the G72 protein interacts physically with DAAO (Reference Chumakov, Blumenfeld and Guerassimenko39). Subsequent meta-analysis found highly significant association between nucleotide variations in the G72/G30 region and schizophrenia (Reference Detera-Wadleigh and McMahon41).
Klein and Kamin (Reference Klein and Kamin42) first reported on sodium benzoate (SB) as a prototype competitive DAAO inhibitor (Ki≈16 μM), as early as in the 1940s (Reference Ferraris, Duvall and Ko43). More recently, Lane et al. (Reference Lane, Lin and Green44) performed a randomised, double-blind, placebo-controlled study using SB in stabilised patients with schizophrenia. Given at a dose of 1 g/day for 6 weeks, SB produced a 21% improvement in Positive and Negative Syndrome Scale (PANSS) total scores and large effect sizes in the PANSS total and subscales, Scales for the Assessment of Negative Symptoms (SANSS)-20 items, Global Assessment of Function, Quality of Life Scale, and Clinical Global Impression, as well as improved neurocognition (Reference Lane, Lin and Green44). In addition, SB was well tolerated without significant adverse effects. However, there are no reports demonstrating the antipsychotic effects of SB in animal models of schizophrenia, although SB could be a potential therapeutic drug for this disorder.
In the present study, we examined whether SB attenuated pre-pulse inhibition (PPI) deficits and hyperlocomotion in mice after the administration of PCP. In addition, we measured levels of d-serine in the blood and in brain regions after oral administration of SB. We also measured levels of other the amino acids, l-serine, glycine, glutamate, glutamine, and γ-aminobutyric acid (GABA), as they are involved in the glutamine–glutamate–GABA cycle (Reference Hashimoto9,Reference Hashimoto, Engberg, Shimizu, Nordin, Lindstrom and Iyo45,Reference Hashimoto46).
Materials and methods
Animals
Male ddY mice (8 weeks old) weighing 25–30 g were purchased from SLC Japan (Hamamatsu, Shizuoka, Japan). The mice were housed in clear polycarbonate cages (22.5×33.8×14.0 cm) in groups of five or six per cage under a controlled 12/12 h light–dark cycle (lights on from 07:00 a.m. to 07:00 p.m.), with room temperature at 23±1°C and humidity at 55±5%. The mice were given free access to water and food pellets. The experimental procedure was approved by the Animal Care and Use Committee of Chiba University.
Drugs
Sodium benzoate (SB; Wako Pure Chemical Co., Tokyo, Japan) was dissolved in 0.5% carboxymethy cellulose (CMC) (Wako Pure Chemical Co.). PCP hydrochloride was synthesised in our laboratory, and the dose (3.0 mg/kg) of PCP was expressed as a hydrochloride salt (Reference Shirai, Fujita and Hashimoto22). L-701,324 (Sigma-Aldrich Co., Ltd., St Louis, MO, USA) was dissolved in 20% polyethylene glycol (PEG300; Wako Pure Chemical Co.) with pH adjusted to 10 with 1 M NaOH. Other drugs were purchased from commercial sources.
Effect of SB on PPI deficits after a single administration of PCP
The mice were tested for their acoustic startle reactivity in a startle chamber (SR-LAB; San Diego Instruments, San Diego, CA, USA) using the standard methods described previously (Reference Shirai, Fujita and Hashimoto22,Reference Zhang, Shirayama, Iyo and Hashimoto47–Reference Ren, Zhang, Fujita, Ma, Wu and Hashimoto50). The test sessions were started after an initial 10-min acclimation period in the chamber. The mice were subjected to one of the following six trials: (1) pulse alone, as a 40 ms broadband burst; a pulse (40 ms broadband burst) preceded by 100 ms with a 20 ms pre-pulse that was (2) 4 dB, (3) 8 dB, (4) 12 dB, or (5) 16 dB over background (65 dB); and (6) background only (no stimulus). The amount of pre-pulse inhibition (PPI) was expressed as the percentage decrease in the amplitude of the startle reactivity caused by presentation of the pre-pulse (% PPI). SB (30, 100, or 1000 mg/kg) or vehicle (0.5% CMC) (10 ml/kg) was administered orally 60 min (including the 10 min acclimation period) before the machine records, and PCP (3.0 mg/kg) or saline (10 ml/kg) was administered subcutaneously (s.c.) 10 min (including the 10 min acclimation period) before. The PPI test lasted 20 min in total.
Effect of SB and L-701,324 on PPI deficits after a single administration of PCP
In order to study the role of the glycine site of the NMDA receptor, we examined the effects of L-701,324, an antagonist of the glycine site of the NMDA receptor, on the effect of SB on PCP-induced PPI deficits in mice. Thirty minutes after oral administration of SB (1000 mg/kg) or vehicle (0.5% CMC) (10 ml/kg), L-701,324 (10 mg/kg) or vehicle (20% PEG) was administered intraperitoneally (i.p.) 30 min later. Thirty minutes after the injection of L-701,324 (or vehicle), PCP (3.0 mg/kg) or saline (10 ml/kg) was administered s.c. The PPI test was performed as described above.
Effect of SB on hyperlocomotion after a single administration of PCP
After habituation (30 min) in the cage, SB or vehicle was injected into the mice (each group n=8–12). One hour after a single oral administration of SB (1000 mg/kg) or vehicle (10 ml/kg, 0.5% CMC), PCP (3.0 mg/kg) or vehicle (physiological saline; 10 ml/kg) was administered s.c. into the mice. Locomotor activity was measured using an animal movement analysis system (SCANET MV-40; Melquest, Toyama, Japan). The system consisted of a rectangular enclosure (560×560 mm). The side walls (height, 60 mm) of the enclosure were equipped with 144 pairs of photosensors located at 6-mm intervals at a height of 30 mm from the bottom edge. An animal was placed in the observation cage 30 min (habituation) before injection of vehicle or SB. Vehicle or PCP was injected 60 min after oral injection of vehicle or SB, and the locomotion activity was measured for 60 min after injection of vehicle or PCP. A pair of photosensors was scanned every 0.1 s to detect the animal’s movements. The intersection of paired photosensors (10 mm apart) in the enclosure was counted as one unit of locomotor activity. Data collected for total 150 min were used in this study. The sum of locomotion in mice for 60 min after the PCP administration was used for data analysis.
Measurement of amino acids by high-performance liquid chromatography (HPLC)
One hour after the single oral administration of SB (1000 mg/kg), mice were killed by decapitation after collecting blood samples. The brain was removed and the frontal cortex, hippocampus, and striatum were dissected on ice. Plasma and brain tissues were frozen on dry ice and stored at −80°C until analysis.
In brief, plasma (20 µl) was homogenised in 180 µl of methanol (HPLC grade) on ice. The homogenates were centrifuged at 3000× g for 6 min at 4°C, and 20 µl of the supernatant was evaporated to dryness at 40°C. To the residue, 20 µl of H2O (HPLC grade), 20 μl of 0.1 M borate buffer (pH 8.0), and 60 μl of 50 mM 4-fluoro-7-nitro-2,1,3-benzoxadiazole (NBD-F; Tokyo Kasei Kogyo Co., Ltd., Tokyo, Japan) in CH3CN (HPLC grade) were added. The reaction mixture was then heated at 60°C for 2 min, and was immediately supplemented with 100 μl of H2O/CH3CN (90/10) containing 0.1% trifluoroacetic acid to stop the reaction. Brain tissues were homogenised in 1.5 ml of methanol (HPLC grade) on ice. The homogenates were centrifuged at 3000× g for 6 min at 4°C, and 20 μl of the supernatant was evaporated to dryness at 40°C. To the residue, 20 μl of H2O (HPLC grade), 20 μl of 0.1 M borate buffer (pH 8.0), and 60 μl of 50 mM NBD-F in CH3CN (HPLC grade) were added. The reaction mixture was then heated to 60°C for 2 min, and was immediately supplemented with 100 μl of H2O/acetonitrile (90/10) containing 0.1% trifluoroacetic acid (TFA) to stop the reaction. Levels of amino acids (d-serine, l-serine, glycine, glutamine, glutamate, and GABA) were measured using high-performance liquid chromatography (HPLC) system (Shimadzu Corporation, Kyoto, Japan), as previously reported (Reference Fukushima, Kawai, Imai and Toyo’oka51–Reference Horio, Kohno and Fujita53). Fluorescence detection was performed at 530 nm with an excitation wavelength of 470 nm.
Statistical analysis
The data are presented as the mean±standard error of the mean (SEM). The PPI data were analysed by multivariate analysis of variance (MANOVA), followed by post-hoc Fisher’s Least Significance Difference (LSD) test. The data of hyperlocomotion were analysed by one-way analysis of variance (ANOVA), followed by post-hoc Fisher LSD test. The data of amino acids were analysed using the Student t-test. Significance for the results was set at p<0.05.
Results
Figure 1 shows the effects of SB (100, 300, or 1000 mg/kg) on PCP (3.0 mg/kg)-induced PPI deficits in mice. The MANOVA analysis of all PPI data revealed that there was a significant effect (Wilks lambda=0.346, p<0.001). Subsequent ANOVA analysis revealed the significant differences (p<0.001) at all dB groups (69, 73, 77, and 81 dB). A post-hoc analysis indicated a significant (p<0.001) difference in PPI deficits between the vehicle+vehicle group and vehicle+PCP (3.0 mg/kg) group at all dB groups (Fig. 1). Pre-treatment with SB (100, 300, or 1000 mg/kg) attenuated PCP-induced PPI deficits in a dose-dependent manner. High dose (1000 mg/kg) of SB significantly (p<0.001) attenuated PCP-induced PPI deficits at all dB groups (Fig. 1). Moderate dose (300 mg/kg) of SB significantly (p<0.05 at 69–77 dB groups, p<0.001 at 81 dB group) attenuated PCP-induced PPI deficits at all dB groups (Fig. 1). In contrast, PPI in mice after administration of SB (1000 mg/kg) alone was similar to control mice (Fig. 1).

Fig. 1 Effect of sodium benzoate (SB) on phencyclidine (PCP)-induced pre-pulse inhibition (PPI) deficits in mice. One hour after the single oral administration of vehicle (10 ml/kg) or SB (100, 300, or 1000 mg/kg), PCP (3 mg/kg) or saline (10 ml/kg) was administered subcutaneously (s.c.) to the mice. Each value is the mean±SEM (n=17–21 per group). *p<0.05, ***p<0.001 as compared with the vehicle+PCP-treated group.
In order to study the role of the glycine site of the NMDA receptor, we examined the effect of L-701,324, an antagonist at the glycine site of the NMDA receptor, on the effect of SB on PCP-induced PPI deficits. Figure 2 shows the effects of SB (1000 mg/kg) and L-701,324 (10 mg/kg) on PCP (3.0 mg/kg)-induced PPI deficits in mice. The MANOVA analysis of all PPI data revealed that there was a significant effect (Wilks lambda=0.193, p<0.001). A post-hoc analysis indicated a significant (p<0.001) difference in PPI deficits between the vehicle+vehicle group and vehicle+PCP (3.0 mg/kg) group at all dB groups (Fig. 2). Pre-treatment with SB (1000 mg/kg) significantly attenuated PCP-induced PPI deficits. However, L-701,324 (10 mg/kg) did not affect the effect of SB on PCP-induced PPI deficits (Fig. 2). Furthermore, L-701,324 (10 mg/kg) did not affect PCP-induced PPI deficits in mice (Fig. 2).

Fig. 2 Effects of sodium benzoate (SB) and L-701,324 on phencyclidine (PCP)-induced pre-pulse inhibition (PPI) deficits in mice. Thirty minutes after the single oral administration of vehicle (10 ml/kg) or SB (1000 mg/kg), L-701,304 (10 mg/kg) or vehicle (10 ml/kg) was administered intraperitoneally (i.p.) to the mice. Thirty minutes after i.p. injection of L-701,304 (or vehicle), PCP (3 mg/kg) or saline (10 ml/kg) was administered subcutaneously (s.c.) to the mice. Each value is the mean±SEM (n=8–11 per group). *p<0.05, **p<0.01, ***p<0.001 as compared with the vehicle+PCP-treated group.
A single administration of PCP (3.0 mg/kg, s.c.) markedly increased locomotion in mice. One-way ANOVA revealed significant differences among the four groups [F(3, 35)=6.17, p=0.002]. Pre-treatment with SB (1000 mg/kg) significantly (p<0.01) attenuated PCP-induced hyperlocomotion in mice (Fig. 2). In contrast, administration of SB (1000 mg/kg) alone did not affect spontaneous locomotion in mice.
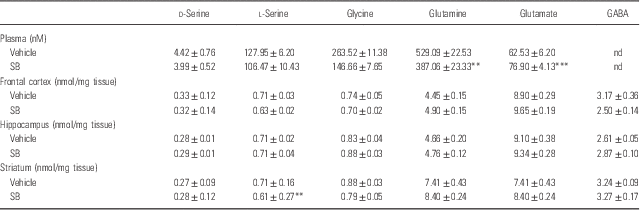
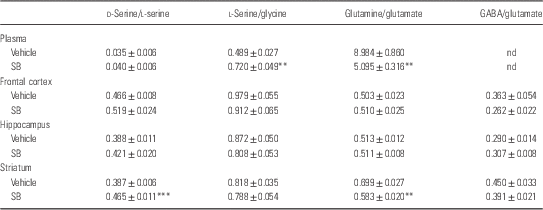
A single oral administration of SB (1000 mg/kg) did not alter plasma levels of d-serine, l-serine, and glycine. In contrast, SB significantly decreased plasma levels of glutamine, whereas SB significantly increased plasma levels of glutamate (Table 1). Furthermore, SB significantly increased the ratio of l-serine to glycine in plasma, suggesting that SB may affect the l-serine–glycine cycle (Table 2). Moreover, SB significantly decreased the ratio of glutamine to glutamate in plasma, suggesting that SB may affect the glutamine–glutamate cycle (Table 2).
Table 1 Levels of amino acids in the plasma, frontal cortex, hippocampus, and striatum 1 h after a single oral administration of vehicle and sodium benzoate (SB: 1000 mg/kg)
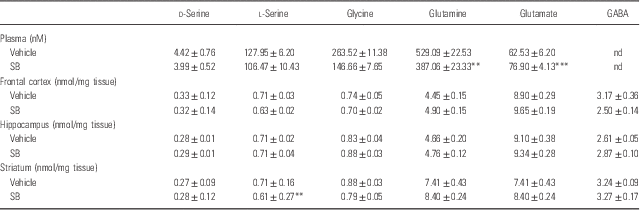
ND, not determined.
Data are expressed as the mean±SEM (vehicle: n=8, SB: n=8).
**p<0.01, ***p<0.001 compared with vehicle-treated group (Student’s t-test).
Table 2 Ratios of amino acid levels in the plasma, frontal cortex, hippocampus, and striatum 1 h after a single oral administration of vehicle and sodium benzoate (SB: 1000 mg/kg)
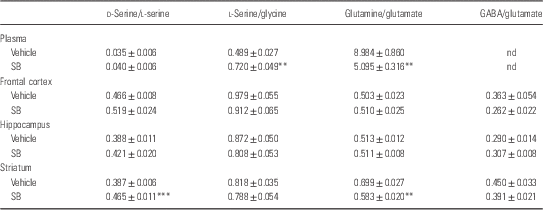
ND, not determined.
Data are expressed as the mean±SEM (Control: n=8, SB: n=8).
**p<0.01, ***p<0.001 compared with vehicle-treated group (Student’s t-test).
A single oral administration of SB (1000 mg/kg) did not alter tissue levels of d-serine and other amino acids except l-serine levels in the striatum (Table 1). However, SB significantly increased the ratio of d-serine to l-serine in the striatum, but not in the frontal cortex and hippocampus. Furthermore, SB significantly decreased the ratio of glutamine to glutamate in the striatum, but not in the frontal cortex and hippocampus. These findings suggest that SB may affect d-serine–l-serine cycle and glutamine–glutamate cycle in the striatum (Table 2).
Discussion
In this study, we found that SB attenuated PPI deficits and hyperlocomotion in mice after the administration of PCP. Furthermore, L-701,324 did not affect the effect of SB on PCP-induced PPI deficits, suggesting that activation at the glycine site of the NMDA receptor may not be involved in the mechanism of action of SB. This is the first report to demonstrate that SB is effective in the PCP model of schizophrenia. However, SB (1000 mg/kg) did not increase the tissue levels of d-serine in the mouse brain, indicating that d-serine in the brain may not be involved in the acute therapeutic action of SB in this model. In contrast, a single dose of SB significantly increased the ratio of d-serine to l-serine in the striatum, suggesting that SB may affect the d-serine–l-serine cycle. Therefore, it is likely that repeated administration of SB increases d-serine levels in the brain, although a further study is needed to confirm this.
Although DAAO inhibitors were proposed as new therapeutic drugs for schizophrenia, their clinical use has been largely unsuccessful (Reference Smith, Uslaner and Hutson54,Reference Sacchi, Rosini, Pollegioni and Molla55). Ferraris et al. (Reference Ferraris, Duvall and Ko43) reported 5-chloro-benzo[d]isoxazol-3-ol (CBIO; IC50=1680 nM) as being a more potent DAAO inhibitor than SB (Ki≈16 μM). In a subsequent report, we found that a single oral dose of CBIO (30 mg/kg) did not increase levels of d-serine in the plasma or in the frontal cortex, and that CBIO alone did not improve the NMDA receptor antagonist dizocilpine-induced PPI deficits in mice (Reference Hashimoto, Fujita and Horio48). In addition, we found that a low dose of d-serine (30 mg/kg) did not improve dizocilpine-induced PPI deficits in mice, although this dose significantly increased plasma levels of d-serine (Reference Hashimoto, Fujita and Horio48). Taken together, it is likely that the extensive inhibition of DAAO in the periphery and brain has a limited effect on brain or extracellular levels of d-serine, and that the behavioural effects of DAAO inhibitors may be very weak. In contrast, we found that co-administration of CBIO with d-serine (or d-alanine) increased levels of D-serine in the brain compared with d-serine (or d-alanine) alone, and that CBIO potentiated the effects of d-serine (or d-alanine) on dizocilpine-induced PPI deficits in mice (Reference Ferraris, Duvall and Ko43,Reference Hashimoto, Fujita and Horio48,Reference Horio, Fujita and Ishima49). Therefore, we proposed that combination therapy of d-serine (or d-alanine) with a DAAO inhibitor could reduce doses of d-serine (or d-alanine) in humans, particularly because the clinical doses of d-serine (or d-alanine) are quite high (30–60 mg/kg) (Reference Ferraris, Duvall and Ko43,Reference Hashimoto, Fujita and Horio48,Reference Horio, Fujita and Ishima49).
DAAO exhibits very low activity in adult forebrains, with high activity in the adult cerebellum. Therefore, it is possible that this increase in cerebellar d-serine levels by DAAO inhibition may, in part, confer antipsychotic effects by augmenting d-serine-mediated regulation of NMDA receptors in the cerebellum (Reference Hashimoto9,Reference Hashimoto56), although we did not measure these levels in the present study. Recent reports show that SB upregulated brain-derived neurotrophic factor (BDNF) in mice (Reference Hashimoto9,Reference Jana, Modi, Roy, Anderson, van Breemen and Pahan57). This implies that the therapeutic effect of SB may be mediated through increased BDNF levels, as the TrkB agonist, 7,8-dihydroxyflavone, attenuated the behavioural abnormalities of hyperlocomotion and PPI deficits in mice after administration of the stimulant methamphetamine (Reference Ren, Zhang, Fujita, Ma, Wu and Hashimoto50,Reference Ren, Zhang, Ma, Fujita, Wu and Hashimoto58).
The glutamine–glutamate cycle in the glia–neuron communication is involved in the glutamatergic neurotransmission in the brain (Reference Hashimoto, Shimizu and Iyo6,Reference Hashimoto, Engberg, Shimizu, Nordin, Lindstrom and Iyo45,Reference Hashimoto46). In this study, we found that SB significantly increased the ratio of glutamine to glutamate, a marker for the glutamine–glutamate cycle, in the plasma and striatum. These findings suggest that SB can affect the glutamine–glutamate cycle in the striatum and plasma, resulting in the regulation of the NMDA receptor.
Accumulating evidence suggests a role for inflammation and oxidative stress in the pathophysiology of schizophrenia (Reference Hashimoto59–Reference Steullet, Cabungcal and Monin63). SB is thought to have a potent anti-inflammatory effect via modulation of the mevalonate pathway and p21ras (Reference Brahmachari, Jana and Pahan64). In addition, SB upregulates the neuroprotective protein, DJ-1, a Parkinson disease protein, also via the modulation of the mevalonate pathway (Reference Khasnavis and Pahan65). Previously, we reported that potent anti-inflammatory molecules and antioxidants, including minocycline and sulphoraphane, attenuate behavioural abnormalities in mice after administration of PCP or methamphetamine (Reference Fujita, Ishima and Kunitachi21,Reference Shirai, Fujita and Hashimoto22,Reference Zhang, Shirayama, Iyo and Hashimoto47,Reference Zhang, Kitaichi and Fujimoto66,Reference Chen, Wu and Zhang67). Taken together, it is possible that SB mediates its therapeutic action through anti-inflammatory and antioxidant pathways. Further detailed studies on molecular targets of SB are needed.
Conclusions
Our study suggests that SB shows potential anti-psychotic activity in animal models of schizophrenia. It is possible that SB could be used for the effective and safe treatment of schizophrenia, particularly because SB is generally recognised as a safe food preservative. In addition, the use of amino acids including d-serine as biomarkers for treatment efficacy will be an interesting future development.

Fig. 3 Effect of sodium benzoate (SB) on phencyclidine (PCP)-induced hyperlocomotion in mice. One hour after the single oral administration of vehicle (10 ml/kg) or SB (1000 mg/kg), PCP (3.0 mg/kg) or saline (10 ml/kg) was administered subcutaneously (s.c.) into the mice. Behaviour (locomotion) in the mice was evaluated for 1 h after administration of PCP. Each value is the mean±SEM (n=8–12 per group). **p<0.01, ***p<0.001 as compared with the vehicle+PCP group.
Acknowledgements
A.M for substantial contributions to conception and design, acquisition of data, analysis and interpretation of the data, drafting the article, final approval of the version to be published; Y.F. for substantial contributions to conception and design, final approval of the version to be published; M. I. for substantial contributions to conception and design, final approval of the version to be published; K.H. for substantial contributions to conception and design, acquisition of data, analysis and interpretation of the data, drafting the article, final approval of the version to be published.
Financial Support
This study was supported by a Grant-in-Aid for Scientific Research on Innovative Areas of the Ministry of Education, Culture, Sports, Science and Technology, Japan (to K.H.).
Ethical Standards
The experimental procedure was approved by the Animal Care and Use Committee of Chiba University. The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional guides on the care and use of laboratory animals.