In several diseases, a large increase in venous admixture in the pulmonary venous return causes cyanosis that may be very severe. These diseases include:
-
1. Congenital heart lesions after partial or complete cavopulmonary anastomosis from Glenn, Kawashima, and Fontan–Kreutzer operations.
-
2. Rarely in congenital heart disease without previous surgery.
-
3. Pulmonary arteriovenous fistula without other congenital heart diseases, often associated with hereditary haemorrhagic telangiectasia (Osler–Rendu–Weber syndrome).
-
4. Certain types of liver disease – the hepatopulmonary syndrome.
An important question is whether these disorders have any causal mechanisms in common.
In this review, the normal venous admixture is described, and then the pathological entities are discussed.
Normal subjects
Intrapulmonary shunting
The oxygen tension is always higher in the alveoli than in the systemic arteries, a difference termed the alveolar–arterial difference. There are several mechanisms for the lower arterial oxygen tension:
-
1. If any blood perfuses inadequately ventilated alveoli, there will be a ventilation–perfusion mismatch and some blood will not be fully oxygenated by those alveoli. Breathing 100% oxygen abolishes this component of the alveolar–arterial difference.Reference Rahn and Fenn 1 , Reference Riley and Cournand 2
-
2. Diffusion limitation causes incomplete oxygenation of the perfused blood. There is no evidence that this can occur normally at rest,Reference Vogiatzis, Zakynthinos and Boushel 3 , Reference Wagner, Gale, Moon, Torre-Bueno, Stolp and Saltzman 4 but it may occur with extreme exerciseReference Hammond, Gale, Kapitan, Ries and Wagner 5 or hepatic cirrhosis.Reference Davis, Schwartz, Lefrak, Susman and Schainker 6 , Reference Schraufnagel and Kay 7
-
3. Anatomic connections allow venous blood to bypass the alveoli. These are termed shunts, and they result in venous admixture that is not affected by breathing 100% oxygen.
-
a. Some of these are post-pulmonary capillary shunts of deoxygenated blood from the Thebesian veins that enter the left atrium or ventricle and deep bronchial veins that drain into pulmonary veins. These shunts are always present, but contribute very little blood flow.
-
b. There may be direct pulmonary artery-to-vein connections that bypass pulmonary capillaries, but these are negligible in the normal subject (see below).
-
Detecting anatomic intrapulmonary shunts
Anatomic shunts may be detected in four ways:
-
1. By intravenous or pulmonary arterial injection of particles of radio-labelled macroaggregated albumin (99mTcMAA) that are too large to pass through normal lung capillaries. Any radioactive counts in peripheral organs such as the kidney or brain indicate bypass of the alveoli – a patent foramen ovale must be excluded – and shunt proportion can be estimated by comparing the counts in the lungs to the counts in the whole body. The method is very sensitive but depends on the size of the particles.
-
2. Contrast echocardiography involves injecting intravenously agitated saline that contains small air bubbles and determining whether any are detected in the left atrium, making sure that they do not pass there via a patent foramen ovale. This method is sensitive but does not give absolute shunt quantity.
-
3. Pulmonary angiography shows either a blush due to excessive numbers of small pulmonary vessels that cannot be distinguished individually or else may show larger tortuous or even aneurysmal connections between the artery and vein. This method is far less sensitive than the two previous methods.Reference Feinstein, Moore, Rosenthal, Puchalski and Brook 8 – Reference Chang, Alejos and Atkinson 10
-
4. By measuring the alveolar–arterial difference while breathing 100% oxygen to eliminate the effects of diffusion and ventilation-perfusion mismatch. This method does not exclude intracardiac right-to-left shunts.
These methods reveal only pulmonary artery-to-pulmonary vein shunts, and do not detect bronchial or Thebesian venous drainage.
Normal intrapulmonary shunts
Normal subjects have a small alveolar–arterial difference of 5–15 mmHg, which is equivalent to <5% of the cardiac output except for neonates in whom it may be more.Reference Nelson, Prod'Hom, Cherry, Lipsitz and Smith 11 Most of this venous admixture is due to the ventilation–perfusion mismatch.
Contrast bubble echocardiography: Pulmonary arteriovenous shunts are rare at rest. In one study, small shunts were found in 0/8 adults while erect but in 2/8 of them while supine,Reference Stickland, Welsh and Haykowsky 12 and another study from the same laboratory found no shunts at rest in 23 normal subjects.Reference Eldridge, Dempsey, Haverkamp, Lovering and Hokanson 13 In both studies, the amount of shunting increased with exercise in parallel with an increased alveolar–arterial difference. All shunting disappeared within 3 minutes of stopping exercise. Another study of 30 young patients after corrective surgery for tetralogy of Fallot showed no shunts at rest.Reference Kim, Bae and Cho 14 Larsson et alReference Larsson, Solymar, Eriksson, de Wahl Granelli and Mellander 9 found no shunting in five normal subjects.
Shunting was observed in 13/13 late-term foetal lambs, 4/4 lambs under 3 days of age, but in 0/7 lambs over 4 weeks of age.Reference McMullan, Hanley, Cohen, Portman and Riemer 15
In normal dogs, microbubbles did not pass through the lungs unless a vasodilator was used or more than 20 ml of saline was injected.Reference Butler and Hills 16
Radiolabelled microspheres: Normal results are given in Table 1.
Table 1 Normal shunting measured by radioactive microspheres.

In dogs, <5% of microspheres 8 μm in diameter passed through the lungs, but occasionally after several circulations even microspheres as large as 50 μm in diameter might pass.Reference Ring, Blum, Kurbatov, Moss and Smith 21 Studies of excised ventilated and perfused lungs of baboons and humans with 25- and 50-μm-diameter microspheres showed <0.2% shunting.Reference Lovering, Stickland, Kelso and Eldridge 22
Respiratory gas techniques: Vogiatzis et alReference Vogiatzis, Zakynthinos and Boushel 3 using Fick techniques calculated that normal subjects during heavy exercise while breathing room air had a maximum of 3.5% (standard deviation (SD) 1.2%) venous admixture that decreased to 0.5% (SD 0.5%) when breathing 100% oxygen.
Anatomy of intrapulmonary shunts
Normal neonates may have small pulmonary arteriovenous connectionsReference Groniowski 23 that may be the remnants of the primitive microvascular plexus in the embryo. These connections exist in adult human lungs as well. In 1965, Elliot and ReidReference Elliott and Reid 24 injected lung vessels and described supernumerary branches of the pulmonary artery that did not accompany airways, branched at right angles from the parent artery, often had a sphincter-like muscle at the origin, and might not communicate with pulmonary capillaries. Similar results were described by others,Reference Tobin 25 , Reference Wilkinson and Fagan 26 and in one of those studies microspheres >50 mm in diameter were found in the pulmonary veins.Reference Wilkinson and Fagan 26
Some studies showed arteriovenous connections subpleurally where alveoli are sparse. It is possible for smaller air bubbles or albumin macroaggregates to pass through the larger capillaries. In rats and dogs, Short et alReference Short, Montoya, Gebb, Presson, Wagner and Capen 27 observed capillary diameters ranging up to 13 μm, with more large capillaries subpleurally, and these figures fit well with an average diameter of 10.2 μm in humans found by scanning electron microscopy.Reference Miura, Nakamura and Matsubara 28 Such connections might explain some of the microparticle passage through normal lungs, but would certainly not explain the alveolar–arterial difference.
Abnormal venous admixture
Cavopulmonary anastomoses
Glenn anastomosis: The Glenn anastomosis is usually the second stage of a planned Fontan–Kreutzer single-ventricle repair. Initially, the right pulmonary artery was transected and anastomosed end to end to the superior caval vein so that blood returning from the upper body was oxygenated in the right lung; this is the classical or unilateral Glenn operation. Return of partly deoxygenated venous blood from the lower body enters the right atrium. It then either bypasses the lungs to enter the systemic circulation through an atrial or ventricular septal defect – tricuspid or pulmonary atresia – or else divides into two streams, one entering the lungs and another, usually larger, passing directly to the systemic circulation. For many years, however, the superior caval vein has been anastomosed end to side to the top of the pulmonary artery so that flows from the upper body are oxygenated in both lungs; this is the bidirectional Glenn procedure, also known as the bidirectional cavopulmonary anastomosis. Depending on the circumstances, the main pulmonary artery may or may not contribute to pulmonary blood flow. There is, however, a variable contribution to superior caval vein flow from the azygos vein that in normal supine adults averages 86–174 ml/min.Reference Lomas, Hayball, Jones, Sims, Allison and Alexander 29 – Reference Bosch, Mastai, Kravetz, Bruix, Rigau and Rodes 31 In most cavopulmonary anastomoses, the azygos vein is ligated at the time of surgery.
Mathur and GlennReference Mathur and Glenn 32 described 5/63 patients who developed pulmonary arteriovenous fistulas after a classical unidirectional Glenn procedure, and McFaul et alReference McFaul, Tajik, Mair, Danielson and Seward 33 described four similar patients. All of these patients had been followed up for many years, and the pulmonary arteriovenous fistulas were confined to the right lung. Trusler et alReference Trusler, Williams and Cohen 34 estimated that 11–21% of patients develop pulmonary arteriovenous fistulas after a classical Glenn operation. With the subsequent use of a connection between the superior caval vein and both pulmonary arteries – bidirectional Glenn anastomosis – the fistulas were found in both lungs.
Kopf et alReference Kopf, Laks, Stansel, Hellenbrand, Kleinman and Talner 35 in a long-term follow-up of 91 patients aged 2 days to 46 years after a Glenn anastomosis observed pulmonary arteriovenous fistulas in 18 (19.7%) of them. A similar study by Cloutier et al,Reference Cloutier, Ash and Smallhorn 19 with an average follow-up of 8.8 years, showed that all 20 patients had increased shunting of radioactive macroaggregates, although only five of these had shunting observed by contrast echocardiography. Feinstein et alReference Feinstein, Moore, Rosenthal, Puchalski and Brook 8 noted angiographically diagnosed fistulas in 66% of lungs sampled. Kim et alReference Kim, Bae and Cho 14 found after an average of 22 months that 19/27 patients developed shunts after the bidirectional Glenn procedure, but only 4/27 showed pulmonary arteriovenous fistulas with angiography. In 14 patients studied 0.1–8.6 years after a bidirectional Glenn shunt for univentricular hearts, 10 (71%) had intrapulmonary shunting by contrast echocardiography but only 3 (21%) showed shunting by angiography.Reference Chang, Alejos and Atkinson 10
Another study observed that 45% of patients had developed shunts after an average of 11.2 years after the operation.Reference Larsson, Solymar, Eriksson, de Wahl Granelli and Mellander 9 There is an increase in the proportion of patients who develop shunts as the follow-up time increases,Reference Trusler, Williams and Cohen 34 , Reference Duncan and Desai 36 although occasionally shunts develop very soon after surgery.
Kawashima operation: In some patients, the intrahepatic portion of the inferior caval vein is absent, because of the failure of the right subcardinal vein to join the hepatic venous system.Reference Chuang, Mena and Hoskins 37 , Reference Mayo, Gray, St Louis, Grosman, McLoughlin and Wise 38 The venous blood returning from the lower body therefore cannot pass directly to the right atrium, but is diverted through the azygos and hemiazygos veins that develop from the supracardinal veins. These pathways drain into the superior caval vein, so that only the hepatic venous and coronary venous returns pass directly into the right atrium. This venous anomaly is particularly common in patients who have left atrial isomerism/polysplenia syndrome, but it can occur with other congenital cardiac anomalies or even in their absence.Reference Debich, Devine and Anderson 39 – Reference Celentano, Malinger, Rotmensch, Gerboni, Wolman and Glezerman 41
In patients with this anomaly, Kawashima and colleagues connected the superior caval vein to the pulmonary arteryReference Kawashima, Kitamura, Matsuda, Shimazaki, Nakano and Hirose 42 , Reference Kawashima, Matsuki, Yagihara and Matsuda 43 and called it the “total cavopulmonary shunt operation”. After this procedure, all systemic venous return except that from the hepatic and coronary veins passes through the superior caval vein to the lungs. Pulmonary arteriovenous fistulas after the Kawashima operation occur in >50% of patients.Reference Brown, Ruzmetov, Vijay, Rodefeld and Turrentine 44 – Reference Setyapranata, Brizard, Konstantinov, Iyengar, Cheung and d'Udekem 48
Congenital heart disease without surgery
Rarely, patients with heterotaxy develop pulmonary arteriovenous fistulas spontaneously.Reference Papagiannis, Kanter and Effman 49 , Reference Gurses, Ulger, Levent and Ozyurek 50 There was one patient who had facial telangiectasia and a cerebral arteriovenous malformation but no family history of hereditary haemorrhagic telangiectasia.Reference Gurses, Ulger, Levent and Ozyurek 50 Srivastava et alReference Srivastava, Preminger and Lock 46 observed such malformations in only 1/56 patients without surgery, and that patient had biliary atresia. Kawata et alReference Kawata, Kishimoto and Ikawa 51 observed pulmonary arteriovenous fistulas in 3/16 patients with left atrial isomerism but in 0/50 with right atrial isomerism.
Miscellaneous forms of congenital heart disease with direct drainage of hepatic veins into the left atrium have also been associated with pulmonary arteriovenous fistulas.Reference Guardado, Byrd and Petersen 40 , Reference Agnoletti, Borghi, Annecchino and Crupi 52 – Reference Stoller, Hoffman, White and Mee 57 There are also patients with isolated drainage of the inferior caval vein into the left atriumReference Al-Ammouri, Shomali and Alsmady 58 – Reference Venables 64 Some of these patients had pulmonary arteriovenous fistulas,Reference Guardado, Byrd and Petersen 40 , Reference Brochard, Lejonc, Loisance and Nitenberg 54 , Reference Stoller, Hoffman, White and Mee 57 , Reference Black, Smith and Goodale 59 some did not,Reference Kloppenburg, Post, Mager and Schepens 56 , Reference Kogon, Fyfe, Butler and Kanter 62 , Reference Meadows, Bergstrand and Sharp 63 and in the others fistulas were not assessed.
Pathology of pulmonary arteriovenous fistulas with congenital heart diseases: One patient developed pulmonary arteriovenous fistulas after a Kawashima operation and had thin-walled pulmonary arterioles and dilated small pulmonary veins, as well as some abnormal thin-walled vessels in the interstitium.Reference Bernstein, Ursell, Brook, Hanley, Silverman and Bristow 65 Two others had dilated terminal bronchial arteries and thickened veins, as well as clusters of thin-walled subpleural vessels.Reference Srivastava, Preminger and Lock 46 One patient with polysplenia but no operation had multiple irregularly shaped vascular channels consistent with pulmonary arteriovenous fistulas.Reference Papagiannis, Kanter and Effman 49
Another two children had excessive small vessels that were thin walled, often dilated, with disordered collagen fibrils and decreased elastic tissue in their walls.Reference Duncan, Kneebone and Chi 66 Some of these vessels appeared to arise from small pulmonary arteries. These abnormal vessels were either dilated (lakes) or were in clusters of small vessels (chains). Some of them appeared to arise from small pulmonary arteries. Immunohistochemistry suggested that the rate of cell proliferation was not increased, and the authors concluded that mechanisms other than vascular proliferation were involved.
In this and a later study,Reference Starnes, Duncan and Kneebone 67 children after a cavopulmonary anastomosis had increased numbers of small vessels on lung biopsy, even in the absence of clinically or angiographically demonstrated pulmonary arteriovenous fistulas. Further investigation of these abnormal vesselsReference Starnes, Duncan and Kneebone 68 showed increased staining for vascular endothelial growth factor and its receptor, and decreased staining for CD31, an indicator of intercellular junctions.
What are these abnormal vessels? We need to distinguish enlargement of pre-existing but inapparent vessels from the budding of new vessels from existing vessels (angiogenesis), and this in turn from vasculogenesis in which angioblasts produce new blood vessels where previously there were none. Vasculogenesis is unlikely because it occurs only with embryonic development and with tumours in tissues devoid of blood vessels. There is evidence for both the other possibilities, and not enough study has been done to determine which predominates.
Isolated pulmonary arteriovenous fistulas
Isolated pulmonary arteriovenous fistulas in the absence of congenital heart disease are uncommon, and are characterised by thin-walled channels lined by endothelium and supported by scanty connective tissue stroma. The fistulas may resemble a plexiform mass of dilated but still small vessels, a tortuous communication between an artery and a vein, or a similar communication with a dilated aneurysmal sac.Reference Sloan and Cooley 69 Rarely, some of these fistulas are present at birth, and are thus truly congenital, but others are detected later and might or might not have been present at birth. There is a strong but not universal association with hereditary haemorrhagic telangiectasia.
Hepatopulmonary syndrome
The hepatopulmonary syndrome is defined as “an arterial oxygenation defect induced by intrapulmonary vascular dilatations (IPVD) associated with hepatic disease”.Reference Rodriguez-Roisin, Krowka, Herve and Fallon 70 , Reference Rodriguez-Roisin and Krowka 71 It is most often due to liver cirrhosis of any cause, with about 15–30% of cirrhotics having the syndrome,Reference Rodriguez-Roisin, Krowka, Herve and Fallon 70 , Reference Gupta, Vijaya and Gupta 72 , Reference Varghese, Ilias-basha, Dhanasekaran, Singh and Venkataraman 73 but any form of acute or chronic liver disease has been associated with hepatopulmonary syndrome. There is a closer association with portal hypertension than with decreased liver function. The lungs contain dilated pre- and post-capillary vessels,Reference Berthelot, Walker, Sherlock and Reid 74 as well as markedly dilated capillaries,Reference Davis, Schwartz, Lefrak, Susman and Schainker 6 , Reference Schraufnagel and Kay 7 so that there is a substantial diffusion defect.Reference Rodriguez-Roisin, Krowka, Herve and Fallon 70 Pulmonary arteriovenous fistulas and ventilation–perfusion mismatch also contribute to the increased venous admixture.Reference Berthelot, Walker, Sherlock and Reid 74 , Reference Agusti, Roca and Rodriguez-Roisin 75 There are also porto-pulmonary anastomoses secondary to the portal hypertension that occurs with most patients with hepatopulmonary syndrome, but these contribute little to the venous admixture.Reference Herve, Lebrec and Brenot 76 Recently, a role for CD68(+) macrophages in the lung as a source of vascular endothelial growth factor and platelet-derived growth factor has been described.Reference Thenappan, Goel and Marsboom 77 , Reference Zhang and Yang 78 These macrophages were found in a cirrhotic model produced by chronic bile duct ligation in rats, and macrophage depletion was shown to prevent or regress the hepatopulmonary syndrome. Similar macrophages were found in autopsies of cirrhotic patients.
Hepatopulmonary syndrome occurs in Budd–Chiari syndrome in which hepatic vein outflow is blocked and the liver is congested; 17/29 patients in one series had some degree of hepatopulmonary syndrome.Reference De, Sen and Biswas 79 The syndrome can occur also with the Abernethy syndrome, in which either the portal vein is absent as a result of which mesenteric blood drains directly into the inferior caval vein or else there is a connection between the portal vein and the inferior caval vein.Reference Alvarez, Ribeiro, Hessel, Baracat and Ribeiro 80 , Reference Howard and Davenport 81 The liver may be normal.
Mechanisms causing pulmonary arteriovenous fistulas: When fistulas were reported after Glenn procedures, a proposed mechanism was the decreased pulsatility in the vessels of the affected lung(s). This hypothesis became untenable when these fistulas did not develop as frequently or even regressed after the complete Fontan–Kreutzer procedure.Reference Srivastava, Preminger and Lock 46 , Reference Knight and Mee 82 Some investigators reported the development of new pulmonary arteriovenous fistulas after the Fontan–Kreutzer operation,Reference Larsson, Solymar, Eriksson, de Wahl Granelli and Mellander 9 , Reference Kwon, Bae, Kim, Noh, Choi and Yun 83 , Reference Moore, Kirby, Madden and Gaither 84 but in many of these patients new fistula development was associated with maldistribution of hepatic venous drainage into the pulmonary arteries, often with disappearance of the fistulas when the flow maldistribution was corrected.Reference Justino, Benson and Freedom 85 – Reference Nakata, Fujimoto and Hirose 89
The hepatic venous drainage was further considered a factor in those patients with congenital drainage of the hepatic veins or inferior caval vein into the left atrium who developed pulmonary arteriovenous fistulas that regressed after restoration of hepatic venous return directly to the pulmonary circulation. Furthermore, in cavopulmonary anastomoses, the hepatic venous drainage reaches the lungs only after passing through the systemic circulation, and re-routing hepatic venous drainage to the pulmonary arteries may cause the venous admixture to decrease and even disappear.Reference Brown, Ruzmetov, Vijay, Rodefeld and Turrentine 44 , Reference Agnoletti, Borghi, Annecchino and Crupi 52 , Reference Knight and Mee 82 , Reference Aidala, Chiappa, Cascarano, Valori and Abbruzzese 90 – Reference Burstein, Mavroudis, Puchalski, Stewart, Blanco and Jacobs 99 Similar changes have been seen after correction of isolated inferior caval vein or hepatic venous drainage into the left atrium.Reference Lee, Menkis and Rosenberg 53 , Reference Brochard, Lejonc, Loisance and Nitenberg 54 , Reference Georghiou, Erez, Bruckheimer, Dagan, Vidne and Birk 100
Dilution or loss of substance?: Owing to the fact that in cavopulmonary anastomosis hepatic venous blood does not pass through the right heart into the lungs, there are two possible mechanisms as causes of the resulting pulmonary arteriovenous fistulas. Either the abnormal pathway for hepatic venous blood dilutes some agonist or else the agonist has a very short half-life. In the normal circulation, about 20–25% of the cardiac output returns to the heart through the hepatic veins,Reference Bradley and Ingelfinger 101 , Reference Wiklund 102 so that in the right heart hepatic venous blood joins the remainder of the venous return and is diluted four- to fivefold by the time it reaches the lungs. After a Kawashima operation, the hepatic venous blood mixes with the remaining venous return in the left heart, and is diluted to the same extent. After a bilateral Glenn operation in which no blood flows through the main pulmonary artery, the hepatic venous blood mixes with the venous return through the inferior caval vein and enters the left heart where it mixes with the blood that has returned through the superior caval vein; the resulting dilution is exactly the same. With both of these shunts, some hepatic venous blood that enters the aorta returns to the lungs via the superior caval vein. The dilution is the same as normal, but only about half the amount of a hepatic factor reaches the lungs in its first passage. This raises the possibility that less of a potential anti-angiogenic factor reaches the lungs in the first passage. However, some first-pass mechanism must be involved, because any long-lasting hepatic factor will eventually reach the lungs with a normal degree of dilution. Therefore, either there is a labile, short-lived hepatic factor that prevents vasodilatation and angiogenesis or else in passage through the systemic circulation angiogenic or vasodilator factors develop and reach the lungs. Both mechanisms might act synergistically in some patients.
Any explanation for the fistulas needs to account for the apparently greater incidence after the Kawashima operation than the conventional bidirectional Glenn operation, and why some patients do not develop pulmonary arteriovenous fistulas with these operations.
Possible mechanisms: Many parts of the body – for example, red and white blood cells, intestinal epithelium, hair root cells, cellular organelles, bone spicules, and myosin – exist in dynamic equilibrium. After some typical time period, the entity breaks down and is replaced. Owing to the fact that the amount of each entity normally remains relatively constant, there must be some feedback system that regulates its quantity. Although such equilibrium has not been studied for pulmonary blood vessels, it is conceivable that the endothelial cells with their exposure to physical forces and chemical activity undergo similar changes. Another type of equilibrium occurs when agonist and antagonist systems maintain the status quo until some trigger mechanism creates an imbalances in the equilibrium; an example of this is the clotting cascade.
A clue to the dynamic factors involved in the pulmonary blood vessels may be found in hereditary haemorrhagic telangiectasia. Several genetic mutations have been associated with hereditary haemorrhagic telangiectasia, the two most prominent involving the endoglin geneReference McAllister, Grogg and Johnson 103 , Reference McAllister, Lennon and Bowles-Biesecker 104 (∼60%) and the activin receptor-like kinase 1 gene (∼30%)Reference Berg, Gallione and Stenzel 105 A rarer mutation (∼4%) occurs in Smad 4.
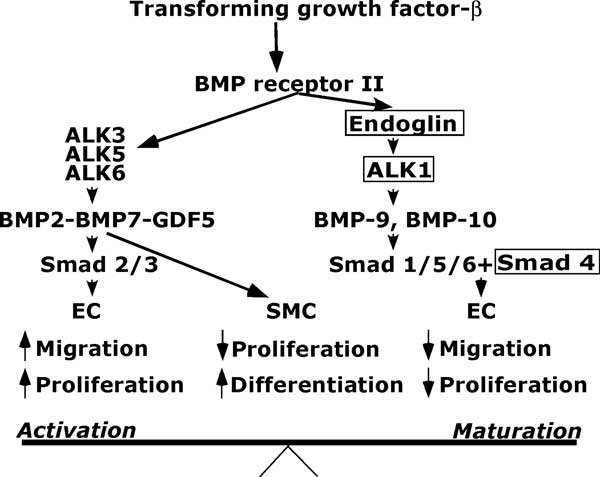
These genes mediate signalling by transforming growth factor-β ligandsReference van den Driesche, Mummery and Westermann 106 , Reference Lebrin, Goumans and Jonker 107 (Fig 1).
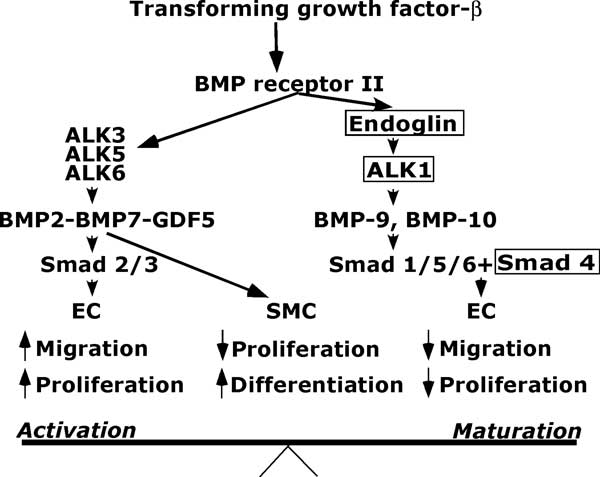
Figure 1 Basic angiogenesis balance. Concepts based on data published by Lebrin et al,Reference Lebrin, Goumans and Jonker 107 David et al,Reference David, Mallet and Keramidas 108 Scharpfenecker et al,Reference Schwarte-Waldhoff and Schmiegel 109 and Carmeliet.Reference Baghdady, Hussein and Shehata 112 EC = endothelial cells; GDF5 = growth differentiation factor; SMC = smooth muscle cells; ALK = activin receptor-like kinase. The boxes indicate sites of known mutation.
There are important interactions among one of the main components of the transforming growth factor-β superfamily, bone morphogenetic protein-9, a circulating protein produced mainly in the liver,Reference David, Mallet and Keramidas 108 activin receptor-like kinase 1, and Smads. Smads are a large family of small messenger proteins that form heterodimers with co-Smad 4 and carry signals from members of the transforming growth factor-β superfamily to the nucleus where they interact with oncogenes, kinases, and other factors to promote or inhibit expression of genes. The affected genes depend on the Smad, cell type, and on the rest of the cell environment.Reference Schwarte-Waldhoff and Schmiegel 109
In a schema described by Lebrin et al,Reference Lebrin, Goumans and Jonker 107 transforming growth factor β acts via transforming growth factor-β receptor II to activate both angiogenic and anti-angiogenic pathways. The angiogenic system includes the activin receptor-like kinase 5-Smad 2/3 pathway that stimulates endothelial cell proliferation and migration. The anti-angiogenic system includes the activin receptor-like kinase 1-Smad 1/5/8 pathway that inhibits endothelial cell proliferation and migration. Endoglin is an accessory transforming growth factor-β receptor that is essential for activin receptor-like kinase 1 signalling and indirectly inhibits activin receptor-like kinase 5 signalling.
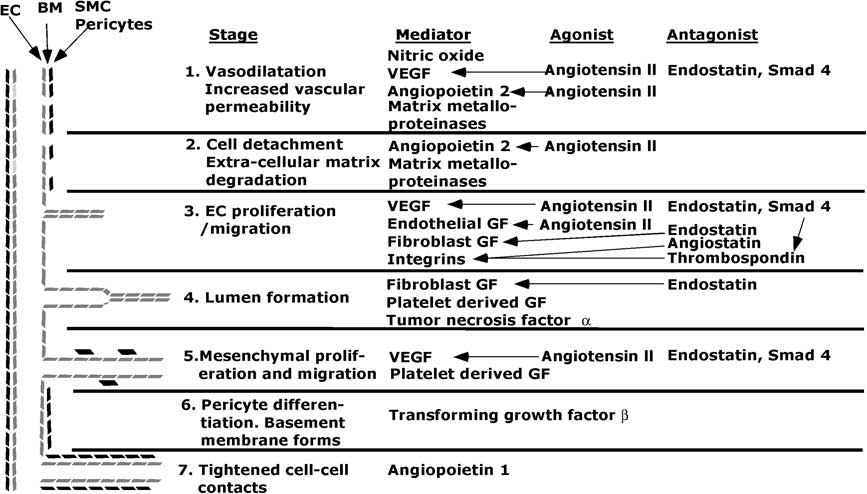
The steps in angiogenesis are beginning to be elucidated (Fig 2).
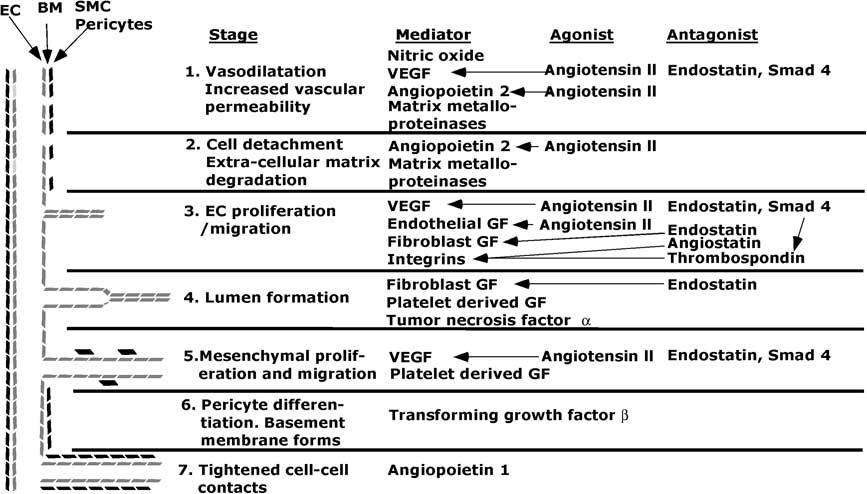
Figure 2 Diagram of stages and some major mediators, agonists and antagonists in angiogenesis. EC = endothelial cells; SMC = smooth muscle cells; BM = basement membrane; VEGF = vascular endothelial growth factor; GF = growth factor. The lines indicate which agonists or antagonists affect a give mediator. In stage 3, Smad 4 increases thrombospondin activity, and thereby increases inhibition of integrins.
The angiogenic process begins with vasodilatation in which nitrous oxide plays a role.Reference Baghdady, Hussein and Shehata 112 – Reference Aydin, Yozgat and Demirkaya 114 Angiopoietin-2 and vascular endothelial growth factor produce fenestrations, increase vascular permeability, and allow plasma proteins to extravasate and, with the aid of cadherins and integrins, form a scaffold for endothelial cell migration. This process is aided by membrane breakdown by activated matrix metalloproteinases. The cords of cells acquire a lumen, and then angiopoietin-1 tightens cell–cell contacts and transforming growth factor-β inhibits endothelial cell proliferation and migration. Periendothelial cells are recruited by platelet-derived growth factor-BB to form smooth muscle cells, and a new basement membrane forms. These processes are held in check by anti-angiogenic factors such as some members of the vascular endothelial growth factor superfamily, thrombospondin-1, and endostatin and angiostatin. Undoubtedly, many other angiogenic and antiangiogenic factors exist and are deployed in various circumstances.Reference Nyberg, Xie and Kalluri 110 , Reference Ribatti 111
Given this array of factors, we should look for one or more factors with angiogenic abilities that reach the lungs in high concentrations only if first-pass hepatic venous drainage bypasses the lungs.
Endoglin per se is not likely to be responsible for the abnormal angiogenesis found in cavopulmonary anastomoses because it is produced widely by endothelial cells. The various isoforms of vascular endothelial growth factor are produced by many types of endothelial cells, and their production is increased by the hypoxia-inducible factor HIF. Their concentrations are increased in children with cyanotic heart disease,Reference Ribatti 111 and there is usually an inverse relation between vascular endothelial growth factor concentration and oxygen saturation.Reference Baghdady, Hussein and Shehata 112 – Reference Suda, Matsumura, Miyanish, Uehara, Sugita and Matsumoto 118 In one study of cyanotic children, not only did vascular endothelial growth factor and basic fibroblast growth factor have higher concentrations in cyanotic than acyanotic children, but in the cyanotic children vascular endothelial growth factor concentrations were considerably higher in the superior caval vein (mean 136 pg/ml) than the inferior caval vein (mean ∼75 pg/ml).Reference Starnes, Duncan and Kneebone 117 In all, 6/22 of these cyanotic patients had had a bidirectional Glenn operation, but unfortunately their results were not separated from the others. Nevertheless, these findings suggest an added layer of complexity in understanding the mechanisms underlying these pulmonary arteriovenous fistulas.
The angiopoietins, like the vascular endothelial growth factor family, are glycoproteins. Angiopoietin-1 is produced by mesenchymal cells, particularly smooth muscle cells. Angiopoietin-2 is produced by endothelial cells in many organs.
Liver factors that might affect angiogenesis include endostatin and angiostatin, both inhibitors of angiogenesis, and angiotensin that can stimulate angiogenesis. Therefore, a deficiency of the first two agents or a surplus of the third might play a role in the formation of pulmonary arteriovenous fistulas in liver disease or diversion of hepatic blood from first passage through the lung.
Endostatin is the C-terminal fragment of collagen XVIII, a key constituent of basement membrane.Reference Seppinen and Pihlajaniemi 119 , Reference Zheng 120 Collagen XVIII has two isoforms – a short form with 303 residues produced by stellate and endothelial cells in the liver that is attached to the basement membrane, and a long form with 493 residues that is produced by hepatocytes and circulates as a plasma protein.Reference Clement, Musso, Lietard and Theret 121 , Reference Musso, Theret and Heljasvaara 122 Endostatin is produced when the tumour suppressor p53 upregulates the activity of a (II) collagen prolyl-4-hydroxylaseReference Teodoro, Parker, Zhu and Green 123 , Reference Lee, Tjin Tham Sjin and Movahedi 124 Although it meets some of the requirements as a causative factor in pulmonary arteriovenous fistulas, endostatin seems to have its major effect on pathologically induced angiogenesis, as in cancer,Reference Folkman 125 and has a half-life of about 1–2 hours.Reference Herbst, Hess and Tran 126 , Reference Thomas, Arzoomanian and Alberti 127 Therefore, unless there is some as yet unknown factor associated with the first pass of collagen XVIII through lung, endostatin may not be a primary factor in the formation of these fistulas. Recently, Field-Ridley et alReference Field-Ridley, Heljasvara and Pihlajaniemi 128 observed that after a Glenn procedure in patients endostatin levels decreased from 4.42 to 3.34 ng/ml, whereas collagen XVIII levels were doubled. The findings are of considerable interest, but a relationship to first-pass lung metabolism of collagen XVIII is yet to be demonstrated. p53 activity has been observed in human pulmonary macrovascular endothelial cells.Reference Damico, Simms and Kim 129 It is of interest that collagen XVIII gene expression is upregulated after cavopulmonary anastomosis in the rat.Reference Tipps, Mumtaz, Leahy and Duncan 130
Angiostatin is formed by proteolytic cleavage of plasminogen secreted by the liver.Reference Brauer, Beck, Roderfeld, Roeb and Sedlacek 131 It is one possible missing liver factor, because it is present in hepatic venous blood and has a half-life of about 15 minutes.Reference Gonzalez-Gronow, Grenett, Fuller and Pizzo 132 This, however, might be too long to involve a first-pass mechanism.
An agent that meets some of the requirements is angiotensin. Angiotensinogen, an α-2-globulin formed mainly in the liver, is cleaved by renin to form angiotensin I, a 10-amino-acid peptide with little biological activity. This in turn is cleaved to form the 8-amino-acid peptide angiotensin II by an exopeptidase – angiotensin-converting enzyme – secreted by pulmonary and renal vascular endothelial cells. Angiotensin II is known to induce angiopoietin and vascular endothelial growth factor,Reference Fujiyama, Matsubara and Nozawa 133 and probably stimulates angiogenesis through the angiotensin-2 receptor.Reference Walther, Menrad, Orzechowski, Siemeister, Paul and Schirner 134 Conversely, stimulating the angiotensin-1 receptor decreases angiogenesis. Bone marrow-derived endothelial cells might also be involved.Reference de Resende, Stodola and Greene 135 Angiotensin II is also known to stimulate the production of nitric oxide,Reference Yin, Ma, Zhao, Cheng and Wang 136 possibly via the angiotensin-2 receptors.Reference Walther, Menrad, Orzechowski, Siemeister, Paul and Schirner 134 A similar peptidase termed angiotensin-converting enzyme-2 converts angiotensin II into a heptapeptide angiotensin III that acts via the mas receptor to antagonise almost all the effects of angiotensin II.Reference Grace, Herath, Mak, Burrell and Angus 137 , Reference Clapp, Thebault, Jeziorski and Martinez De La Escalera 138 Therefore, the resultant effect on angiogenesis in the lung will depend in part on the balance of these two angiotensins.
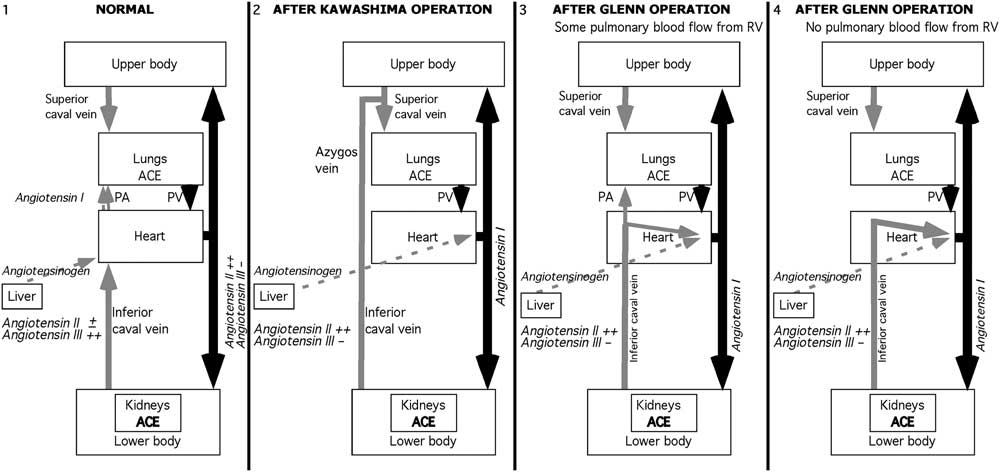
Angiotensin II has a short half-life in the circulation, about 16–30 seconds.Reference Al-Merani, Brooks, Chapman and Munday 139 Therefore, one possible scenario is that with normal hepatic venous drainage into the lung, the angiotensin II that is produced has been partly metabolised to angiotensin III by the time it has passed through the systemic circulation and returned to the lung (Fig 3). Although the difference in time between the appearance of angiotensin II in the aorta and in the renal vein is small, not only is there a small decrease in angiotensin II concentration by the time it reaches the kidney, but there is a substantial uptake of angiotensin II by the kidney that further reduces the concentration of lung-derived angiotensin II in the renal vein.Reference Bailie, Rector and Seldin 140 – Reference Admiraal, Danser, Jong, Pieterman, Derkx and Schalekamp 142

Figure 3 Schema of possible role of angiotensin II in angiogenesis. Panel 1: Normal circulation. Hepatic and lower body venous blood join in the high inferior caval vein to reach the lung. Angiotensin II is produced as blood passes through the lung, but little of it reaches the lung. Panel 2: After a Kawashima operation, angiotensin II is produced in the kidneys and returns rapidly to the lungs via the inferior caval and azygos veins. Panel 3: After a Glenn operation, if some pulmonary blood flow reaches the lungs, then the lungs are exposed to a high concentration of angiotensin II, but in lesser amounts than after a Kawashima operation. Panel 4. If all pulmonary flow is from the superior caval vein, then the concentration of angiotensin II reaching the lung is much lower because it has to circulate first through the upper body. Black arrows = arterial blood. Gray solid arrows = caval venous blood. Gray dashed arrows = hepatic venous blood.
If hepatic venous blood is diverted to the systemic circulation, then angiotensin II is produced as it passes through the renal circulation (Fig 3). After a Kawashima operation, renal venous blood returns rapidly to the lung via the azygos and hemiazygos venous connections to the superior caval vein, so that angiotensin II is present in greater than normal concentrations and angiotensin III in lower concentrations when it reaches the lung. A similar short return pathway from the kidneys occurs after a Glenn operation in which some flow reaches the lung through the main pulmonary artery, as may occur with tricuspid atresia and miscellaneous anomalies including some double-inlet left ventricles and complex anomalies. On the other hand, the azygos vein is usually ligated during the Glenn operation. Therefore, if all pulmonary blood flow after the Glenn operation comes through the superior vena cava, as in pulmonary atresia, hypoplastic left heart syndrome, and many other complex anomalies, then most of the renal venous return reaches the lungs only after returning to the left ventricle, and circulating once more around the upper body before reaching the superior caval vein (Fig 3). In these latter patients, angiotensin has been further metabolised and so is less important as an angiogenic stimulus. Therefore, although angiotensin II is a good candidate for angiogenesis after a Kawashima operation, it may be a poorer candidate after most traditional Glenn operations. However, because angiogenesis is a delicate balance between several angiogenic and anti-angiogenic factors, the balance might easily be upset in more than one way. No matter what the venous return pathway to the lungs is, there will be more angiotensin II and less angiotensin III when hepatic venous blood bypasses the lung in its first circulation.
In one study of children after a bidirectional Glenn operation, those with no other source of pulmonary blood flow had an intrapulmonary shunt averaging 34.9% of cardiac output (SD 15.8%), whereas those with additional flow through the main pulmonary artery had an average shunt of 12.0% (SD 2.6%).Reference Vettukattil, Slavik and Lamb 20 One possible explanation of these findings is that the reduced amount of hepatic blood flow entering the pulmonary artery from the right ventricle contained a reduced amount of a short-lived anti-antiogenic factor, whereas hepatic blood reaching the pulmonary arteries only through the caval anastomosis had even less of this factor. To date, such a short-lived anti-angiogenic factor has not yet been identified. One candidate for this factor might be the incompletely defined anti-angiogenic protein isolated by Marshall et alReference Marshall, Duncan and Jonas 143 who showed that media from cultured rat hepatocytes inhibited the growth of bovine capillary endothelium but not epithelial or tumour cells. The half-life of this protein is unknown. Another candidate is angiotensin III, which also has a short half-life.
The role of angiotensin needs further study. Angiotensin-1 receptors are in greater numbers than angiotensin-2 receptors, and angiotensin-1 receptor activation has variously been reported to promote and inhibit angiogenesis. For example, Amaral et alReference Amaral, Linderman, Morse and Greene 144 , Reference Amaral, Papanek and Greene 145 found that exercise induced angiogenesis in rat limb muscle, and that this action could be blocked by an angiotensin-1 receptor blocker. On the other hand, Walther et alReference Walther, Menrad, Orzechowski, Siemeister, Paul and Schirner 134 did experiments that indicated that the angiotensin-2 receptors but not the angiotensin-1 receptors were involved in angiogenesis. Of more importance is the fact that tissues have a local renin–angiotensin system that might act independently of any circulating agonists.Reference Gwathmey, Alzayadneh, Pendergrass and Chappell 146 , Reference Gwathmey, Westwood and Pirro 147 Nevertheless, there seems enough reason to investigate whether angiotensin is one of the factors promoting angiogenesis in these diseases.
The fact that angiotensin is involved in some way was demonstrated by the finding that after creating a unilateral cavopulmonary anastomosis in sheep the shunted lung had decreases in angiotensin-converting enzyme and angiotensin II, and increases in angiotensin-2 receptors, but these levels returned to normal after 15 weeks. However, angiotensin-1 receptors were persistently upregulated.Reference Malhotra, Reddy and Thelitz 148 , Reference Malhotra, Riemer, Thelitz, He, Hanley and Reddy 149
Bradykinin is an angiogenic factor (among its many actions) that is produced by the action of plasma kallikrein on high molecular weight kininogen that is produced by the liver. It is normally metabolized completely as it passes through the lung into an inert component and a long-acting anti-angiogenic form of high molecular weight kininogen. If bradykinin does not pass through the lung it circulate with a half life of about 30 seconds, possibly long enough to exert an angiogenic effect in the lung.
The role of liver factors has been invoked not only because of the effects of diverting hepatic blood from or returning it to the pulmonary artery, but also because of similarities to the hepatopulmonary syndrome. The comparison should not be pushed too far because of substantial differences between the various disorders. In hepatopulmonary syndrome, for example, the predominant pathology in the lung consists of dilated capillaries that are not a feature of other forms of pulmonary arteriovenous fistulas. The role of macrophages has also not been described other than in the hepatopulmonary syndrome, although their discovery is recent enough that they have not been looked for in other forms of pulmonary arteriovenous fistulas. Then, although in the cardiac forms of pulmonary arteriovenous fistulas there is clear evidence of a first-pass mechanism, in liver disease there must be a constant overproduction of an angiogenic factor or failure of production of an anti-angiogenic factor. There is no need to postulate a labile factor. Angiogenic and vasodilator factors abound in cirrhosis of the liver. There is evidence of overproduction of nitric oxide in cirrhosis. One suggested mechanism for the pathology is stimulation of macrophage production of inducible nitric oxide synthase by tumour necrosis factor-α, and inhibition of tumour necrosis factor-α by pentoxifyllineReference Gupta, Kumar and Jaiswal 150 , Reference Zhang, Ling and Tang 151 decreases nitric oxide production and ameliorates the hepatopulmonary syndrome. Furthermore, patients with cirrhotic livers have increased circulating endotoxinReference Lumsden, Henderson and Kutner 152 that is also vasodilator and angiogenic.Reference Pidgeon, Harmey, Kay, Da Costa, Redmond and Bouchier-Hayes 153 Finally, other characteristics of cirrhosis of the liver such as spider naevi and systemic peripheral vasodilatation are not a feature of other forms of pulmonary arteriovenous fistulas.
Conclusion
The role of hepatic factors in stimulating or inhibiting angiogenesis in the lung appears to require involvement of first-pass metabolism of hepatic venous blood through the lungs. Diversion of hepatic blood from pulmonary capillaries during the first pass implies that there is a short-lived factor that is either anti-angiogenic that bypasses the lungs or else a short-lived pro-angiogenic factor that fails to break down before reaching the lungs in the second passage. Both theories are ripe for further exploration. The fact that in liver disease with the hepatopulmonary syndrome there are also pulmonary arteriovenous fistulas may not be helpful in indicating the cause of pulmonary arteriovenous fistulas after a cavopulmonary anastomosis.
The mechanisms described above do not explain the variability in the timing or extent of pulmonary arteriovenous fistulas in different patients. Whether this is due to variations in the baseline concentrations of various factors involved in angiogenesis, such as vascular endothelial growth factor, or underlying genetic mechanisms is unknown.
Acknowledgement
The author thanks Dr Jeffrey Fineman for allowing him to see his unpublished manuscript, and Dr David Teitel for suggestions about mechanisms.