INTRODUCTION
Larval ecology studies help in understanding the population dynamics, community patterns, ecosystem structure and biodiversity of native and invasive species (Webb et al., Reference Webb, Barnes, Clark and Bowden2006). Identification of marine invertebrate larvae is a tedious, labour intensive task by expert taxonomists. Traditionally, planktonic larval identification is difficult because of larval intricacy and similarity in size, shape and developmental stages (Chanley & Andrews, Reference Chanley and Andrews1971; Branscomb & Vedder, Reference Branscomb and Vedder1982; Shanks, Reference Shanks1986; Nichols & Black, Reference Nichols and Black1994). Due to their small size, shape and similar developmental stages, it is difficult to identify these larvae morphologically, although they play a pivotal role in taxonomic identification (Levin, Reference Levin1990). Sometimes larval identification becomes extremely difficult due to phenotypic plasticity (Hebert, Reference Hebert2002).
Molecular techniques have the potential to accurately identify the organism to its species level, thereby overcoming taxonomic ambiguity. Identification and quantification of marine invertebrate larvae is far easier using molecular techniques (Baldwin et al., Reference Baldwin, Black, Sanjur, Gustafson, Lutz and Vrijenhoek1996; Bilodeau et al., Reference Bilodeau, Lankford, Kim, Felder and Neigel1999; Makinster et al., 1999; Morgan & Rogers, Reference Morgan and Rogers2001; Deagle et al., Reference Deagle, Bax, Hewitt and Patil2003; Larsen et al., Reference Larsen, Frischer, Rasmussen and Hansen2005; Vadopalas et al., Reference Vadopalas, Bouma, Jackels and Friedman2006; Jones et al., Reference Jones, Preston, Marin, Scholin and Vrijenhoek2008; Chen et al., Reference Chen, Hoeg and Chan2013). Polymerase Chain Reaction (PCR) along with sequencing has led to accurate identification of any organism to its species level. Appropriate use of specific primers can facilitate rapid, sensitive and accurate detection of any individual species in a population. Some molecular techniques which assist in identification or characterization of organisms are DNA barcoding (Hebert et al., Reference Hebert, Cywinska, Ball and deWaard2003a, Reference Hebert, Ratnasingham and deWaardb); Random amplified polymorphic DNA (Coffroth & Mulawka, Reference Coffroth and Mulawka1995); multiplex PCR (Hare et al., Reference Hare, Palumbi and Butman2000); Middle repetitive sequence analysis (MaKinster et al., Reference MaKinster, Felder, Chlan, Boudreaux and Neigel1999); Amplified fragment length polymorphism (Bucklin, Reference Bucklin, Harris, Wiebe, Lenz, Skjoldal and Huntley2000; Rogers, Reference Rogers, Atkinson and Thorndyke2001); Restriction fragment length polymorphism and Single strand conformation polymorphism analysis (Hillis et al., Reference Hillis, Mable, Moritz, Hillis, Moritz and Mable1996). Oligonucleotide probes used for specific detection of individual larvae in a mixed population are either concise to family level (Bell & Grassle, Reference Bell and Grassle1998), genus level (Frischer et al., Reference Frischer, Danforth, Tyner, Leverone, Marelli, Arnold and Blake2000) or species level (Frischer et al., Reference Frischer, Danforth, Tyner, Leverone, Marelli, Arnold and Blake2000; Hare et al., Reference Hare, Palumbi and Butman2000). Molecular tools with respect to PCR-based approaches are more reliable and frequently used in larval identification (Hare et al., Reference Hare, Palumbi and Butman2000; Wood et al., Reference Wood, Krogran, Dover, Schneider, Heidt, Boateng, Dean, Golshani, Zhang, Greenblatt, Johnston and Shilatifard2003; Webb et al., Reference Webb, Barnes, Clark and Bowden2006; Chen et al., Reference Chen, Hoeg and Chan2013).
Barnacles are of major concern in biofouling studies around the world. They have drawn the attention of many investigators in marine plankton ecology owing to their easy accessibility on the rocky intertidal regions and also because some species are dominant in marine fouling (Strathmann et al., Reference Strathmann, Branscomb and Vedder1981; Crisp, Reference Crisp, Costlow and Tipper1984; Connell, Reference Connell1985; Holm, Reference Holm1990; Sutherland, Reference Sutherland1990; Bertness et al., Reference Bertness, Gaines, Bermudez and Sandford1991; Raimondi, Reference Raimondi1991; Thiyagarajan et al., Reference Thiyagarajan, Venugopalan, Nair and Subramoniam1997a, Reference Thiyagarajan, Venugopalan, Subramoniam and Nairb). Barnacles possess both a planktotrophic and a lecithotropic larval stage, which settle and metamorphose on hard substratum resulting in macrofouling. Morphological identification of barnacle larval forms in a population is difficult because of their intricacy and similarity in size, shape and developmental stages, and requires extensive microscopy and taxonomic expertise.
Balanus amphitrite, an acorn barnacle, has wide distribution, can be easily maintained in the laboratory, and possesses six planktonic naupliar stages followed by a pre-settlement cypris stage. This species has been extensively used in different studies related to larval development, metamorphosis, influence of different chemical cues and antifouling assays (Rittschof et al., Reference Rittschof, Branscomb and Costlow1984; Maki et al., Reference Maki, Rittschof, Costlow and Mitchell1988; Clare et al., Reference Clare, Freet and McClary1994; Anil et al., Reference Anil, Chiba, Okamoto and Kurokura1995; Khandeparker et al., Reference Khandeparker, Anil and Raghukumar2003, Reference Khandeparker, Anil and Raghukumar2006; Khandeparker & Anil, Reference Khandeparker and Anil2011). Since B. amphitrite larvae are the primary target of investigations related to biofouling and plankton ecology, their fast enumeration and identification is crucial. In the present study, a PCR-based approach was used for detection of a dominant fouling barnacle, B. amphitrite (syn. Amphibalanus amphitrite; Pitombo, Reference Pitombo2004) larvae from the mixed population.
Mitochondrial DNA and nuclear DNA have been the major targets for species identification due to their high conservation and high copy numbers per cell (Stach & Tubeville, Reference Stach and Tubeville2002). Application of mtDNA (12S and 16S) has been useful for species identification, because sequences from various kinds of species have been deposited in the database. Identification of barnacles, based on analysis of 12S and 16S rRNA genes, has been reported by Begum et al. (Reference Begum, Yamaguchi and Watabe2004) and Simon-Blecher et al. (Reference Simon-Blecher, Huchon and Achituv2007). Recently, a species-specific primer for quantitative real-time PCR (qPCR) was evaluated for specific detection and quantification of B. amphitrite using the 12S rRNA gene (Endo et al., Reference Endo, Sato, Matsumura, Yoshimura, Odaka and Nogata2010). Nucleic acid-based sandwich hybridization assays using an rRNA target probe was used for barnacle detection of the group (order Thoracica) and species (Balanus glandula) which could detect even a single barnacle larva in a water column (Goffredi et al., Reference Goffredi, Jones, Scholin, Marin and Vrijenhoek2006). Designing of species-specific primers within the 18S rRNA gene region helps in detecting individual species, since 18S rRNA gene regions have slowly evolved among different orders and families, including invertebrates (Winnepenninckx et al., Reference Winnepenninckx, Backeljau and de Wachter1995; Bleidorn et al., Reference Bleidorn, Vogt and Bartolomaeus2003; Pradillon et al., Reference Pradillon, Schmidt, Peplies and Dubilier2007). In the present study an attempt was made to develop species-specific primers within the 18S rRNA gene region which has not been attempted earlier for B. amphitrite and this provides an additional dimension to this field, especially with reference to identification of B. amphitrite larvae in a mixed zooplankton sample. The primers were designed by comparing the 18S rRNA sequences of closely related Balanus sp. and evaluating the conserved region within the 18S rRNA gene sequence. This approach for planktonic larval detection is less time-consuming compared with morphological microscopic examination and less expensive than other DNA-based approaches.
MATERIALS AND METHODS
Sample collection
Adult B. amphitrite were collected from the intertidal region of Goa, West Coast of India. Adults obtained from field samples were brought to the laboratory and exposed to air for 1–2 h and then immersed in filtered seawater, which triggered the release of larvae. The Instar II nauplius larvae obtained from the adults were used as a positive control and for internal spiking in the present investigation. Horizontal hauls were taken for collection of zooplankton using a 100 µm mesh Heron-Tranter (HT) zooplankton net with a calibrated flow meter attached to it in the viscinity of Dona Paula Bay (15°27.5′N 73°48′E), west coast of India. The plankton samples were either preserved in 95% ethanol or directly processed for DNA isolation and PCR analysis. The preserved samples were quantified for the presence of cirripede larvae. The number of larvae present in different samples varied from 50 to 4000 ind m−3. Four other barnacle species (Chthamalus malayensis, Megabalanus tintinnabulum, Lepas sp. and Ibla sp.) were also collected from the study area. The adult barnacles were identified based on previously described morphological features (Karande, Reference Karande1967; Wagh & Bal, Reference Wagh and Bal1970; Henry & McLaughlin, Reference Henry and McLaughlin1975; Flowerdew, Reference Flowerdew1985; Pitombo, Reference Pitombo2004; Fernando, Reference Fernando2006). They were collected for extraction of genomic DNA and verification of primer specificity.
Extraction of genomic DNA
The adults of B. amphitrite were starved overnight prior to DNA extraction. Genomic DNA was extracted from adult barnacles namely B. amphitrite, Chthamalus malayensis, Megabalanus tintinnabulum, Lepas sp. and Ibla sp. Whole adult muscle tissue was used for genomic DNA isolation. DNA extracted from newly hatched Artemia sp. nauplii was used as control. The zooplankton samples were weighed and then subjected to DNA extraction using DNA Extraction solution (GeneI, India). The extracted DNA was visualized on a 0.8% Agarose gel stained with ethidium bromide and observed under UV illumination.
Designing of B. amphitrite-specific primers
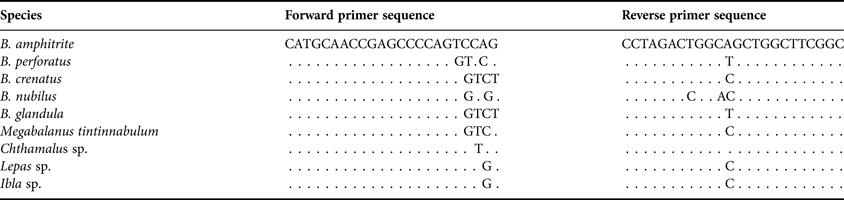
18S rRNA gene sequences of barnacles (Table 1) were obtained from the NCBI GenBank (http://www.ncbi.nim.nih.gov/) and compiled. These 18S rRNA gene sequences were aligned using Clustal X 1.8 (Thompson et al., Reference Thompson, Gibson, Plewnaik, Jeanmougin and Higgins1997). Since the sequence of B. amphitrite 18S rRNA gene was not available in any of the databases, primers were designed by selecting a short highly conserved region within the 5′ end region between barnacles with few mismatches in the last five nucleotides in the 3′ region of the primer (Table 1). Target primers were designed in the gene region where there were mismatch pairs in relation to other barnacle species. Primers were manually designed using BioEdit, with standard priming conditions such as primer length, self annealing, possible loops, GC content and melting temperature (T m), which were evaluated every time during each primer design. During our analysis, some of the 3′ end nucleotides of the forward primer were changed in order to eliminate strong loop, self annealing or primer dimer formation.
Table 1. Balanus amphitrite specific primers aligned with the corresponding sequence from other available barnacles.

A dot represents similar nucleotides within the primer sequence.
Polymerase chain reaction
DNA isolated from the adult samples was used for PCR amplification of the 18S rRNA gene. DNA amplification of the 18S rRNA gene region was performed using primers which annealed only with the B. amphitrite species. 5 µl of extracted DNA was used for PCR amplification using a PTC 200 Thermal cycler (MJ Research). PCR reaction was carried out with a 50 µl reaction mixture containing 10 mM of each dNTPs, 20 pmoles of each primer, 1 U of Taq polymerase, 1× PCR reaction buffer containing 10 mM MgCl2. Amplification of the 18S rRNA gene region was carried out using B. amphitrite specific primers (Table 2). The thermal cycler was programmed using a touchdown PCR protocol. One cycle of 94 °C for 2 min, followed by 35 cycles of 1 min at 94 °C, 45 s at 58 °C, 1 min at 72 °C and final cycle of 10 min at 72 °C. The resulting fragments were resolved electrophoretically on 1% agarose gel for 1 h at 90 V. The resulting amplicons were compared with a commercial 1 Kb DNA ladder (Genetix, India). Amplification was carried out in replicates and batches, to determine the specificity, sensitivity and reproducibility of the designed primers.
Table 2. Sequences of oligonucleotide primers used for species-specific detection of Balanus amphitrite.
Verification of primer's specificity
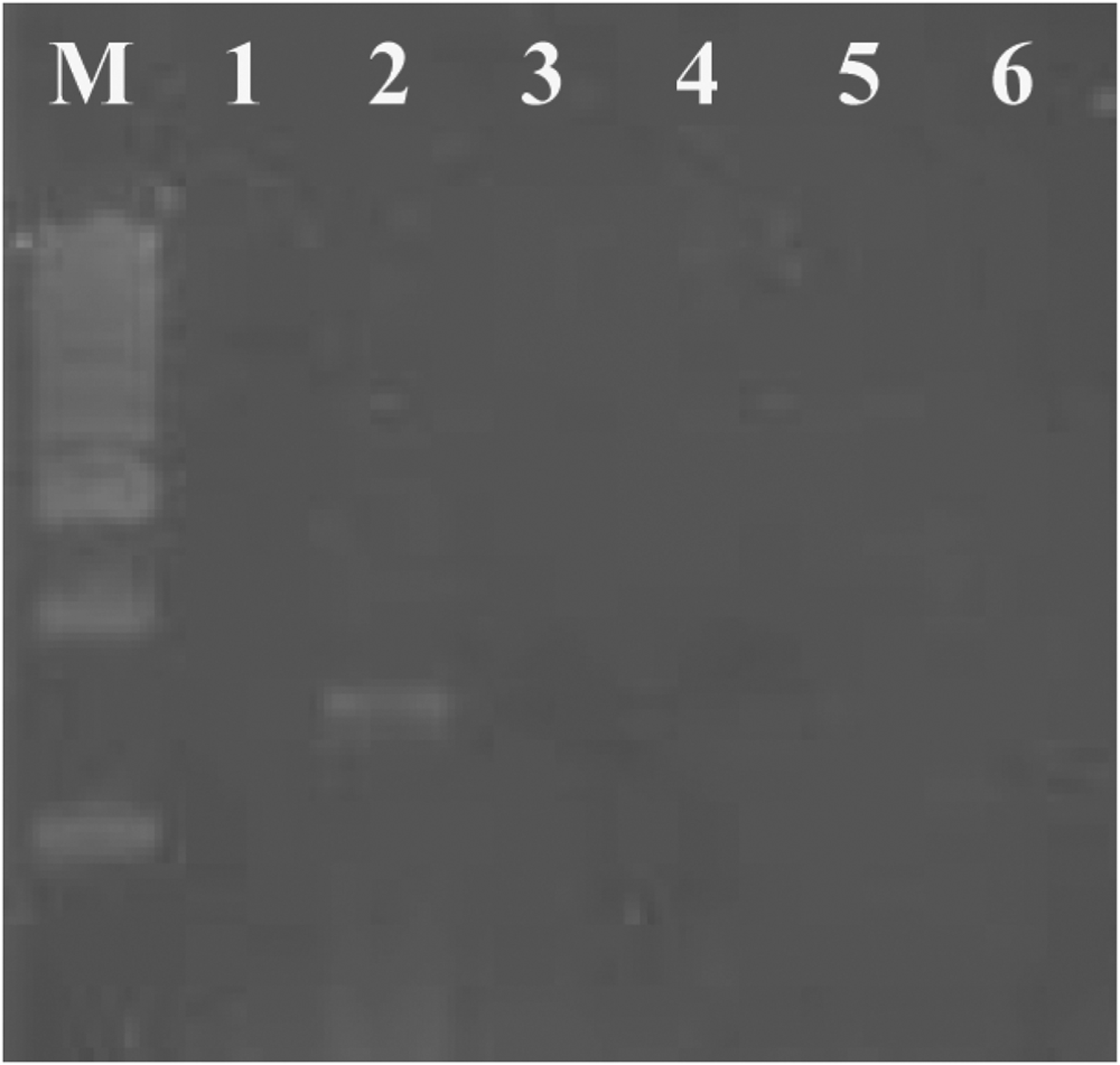
Primer specificity was evaluated by PCR amplification of the 18S rRNA gene region using extracted genomic DNA from four other barnacle species (Chthamalus malayensis, Megabalanus tintinnabulum, Lepas sp. and Ibla sp.). The designed primers amplified ~1600 bp amplicons only from the B. amphitrite (Figure 1). However, other species of barnacles did not show amplification with similar primer and PCR conditions. PCR amplification using gDNA from Artemia sp. also showed no amplicon, resulting as negative control for the designed primers.
Fig. 1. Agarose gel electrophoresis of PCR amplified 1600 bp product of the 18S rRNA gene from Balanus amphitrite using specific primers. Left lane represents standard size markers (1 Kb DNA ladder, Chromous). Lane 1 is control using Artemia sp., Lane 2 is the PCR product of 18S rRNA gene region using B. amphitrite genomic DNA as template. Lanes 3–6 are genomic DNA from other known available barnacles as template: Megabalanus tintinnabulum (3); Chthamalus malayensis (4); Lepas sp. (5); Ibla sp. (6).
Evaluation of primers for B. amphitrite specificity
DNA isolated from mixed zooplankton samples were subjected to PCR amplification using the above protocol. The zooplankton samples were screened under a microscope and all cirripede larvae were eliminated which were used as control, in order to check the specificity of our designed primers. In order to check the presence or absence of any PCR inhibitors in extracted DNA, the zooplankton samples without cirripede larvae were spiked with 10 and 100 larvae (Instar II) of B. amphitrite. Owing to small size and low DNA content in the case of Instar II barnacle nauplii one larva could not be amplified. DNA isolated from Artemia sp. was used as control.
RESULTS AND DISCUSSION
Alignment of the 18S rRNA gene sequences of barnacles (Table 1) obtained from GenBank were evaluated using Clustal X. Sequences of all individuals revealed a high rate of conservation within the 18S rRNA gene region. Very low mismatch regions were present within the 18S rRNA gene region of Balanus sp. The forward primer was designed in this region because it had a conserved 5′ end with few mismatches at the 3′ region and the reverse primer was designed in a conserved region compared with other barnacles (Table 1). The designed primers amplified ~1600 bp amplicons only from the B. amphitrite species (Figure 1).
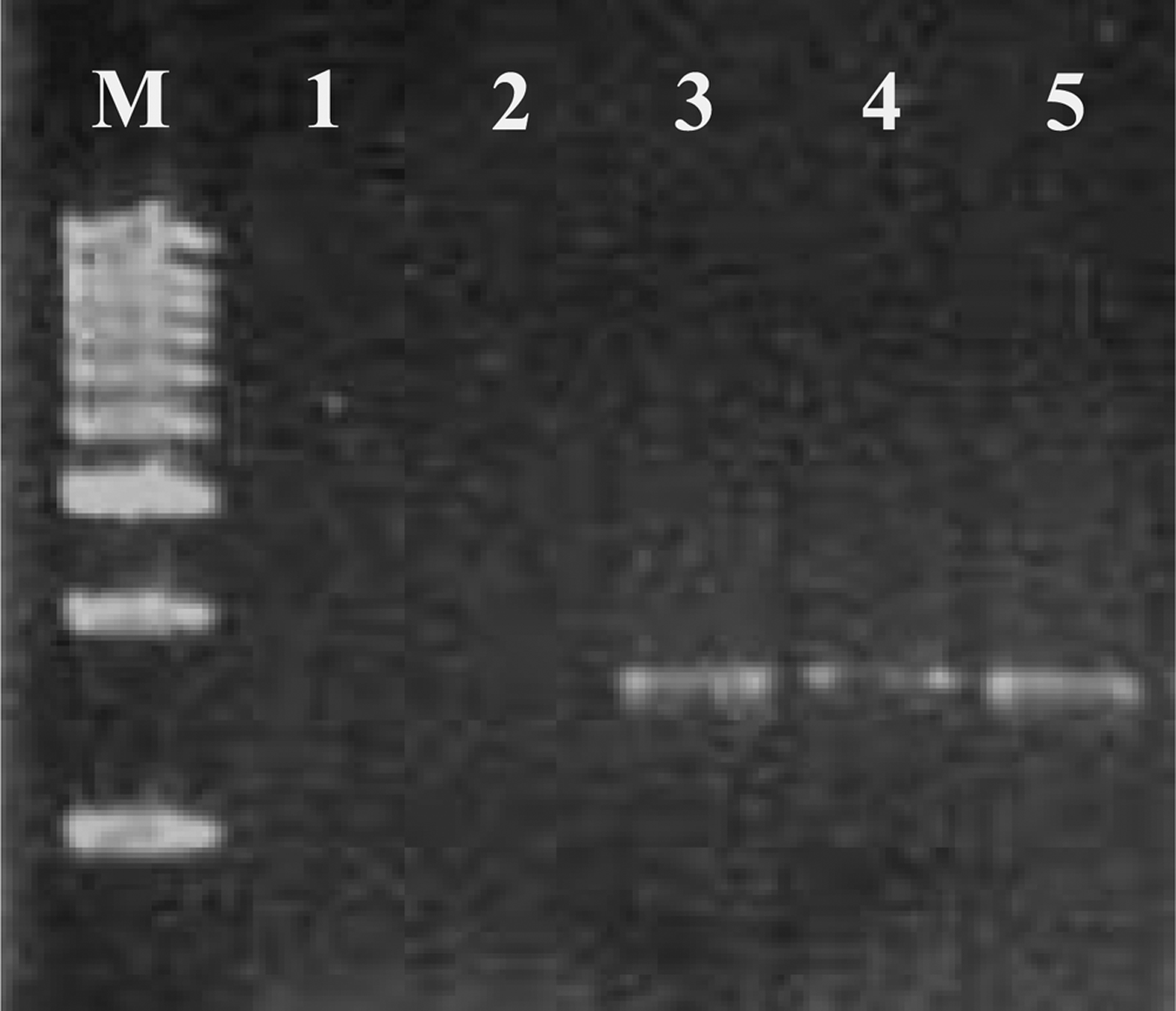
DNA extracted from the mixed planktonic population showed a positive result when amplified with the B. amphitrite specific primer, indicating the presence of B. amphitrite larvae in the mixed sample. The zooplankton samples without cirrepede larvae spiked with 10 and 100 B. amphitrite larvae produced ~1600 bp fragment stating the sensitivity of the designed primer for the specific detection of B. amphitrite and also eliminating the presence of any PCR inhibitors (Figure 2). In the present study a simple and inexpensive methodology was adopted for the specific detection of B. amphitrite larvae in a mixed population. 18S rRNA genes contain regions which are either highly conserved or variable, and specific primers can be targeted to these characteristic sites for families, genus or species (Amann et al., Reference Amann, Krumholz and Stahl1990). Mostly these ribosomal regions are best suited for probe designing (Peplies et al., Reference Peplies, Glockner, Amann and Ludwig2004). In the present study we used this information for designing species-specific primers. However, 28S and the mitochondrial rRNA (12S and 16S rRNA) gene regions can also be used for such studies if there is no single mismatch in the 18S rRNA gene, within the species level. Identification of organisms using a 12S rRNA gene sequence has also been attempted in detection and quantification of barnacle larvae in plankton samples using qPCR (Endo et al., Reference Endo, Sato, Matsumura, Yoshimura, Odaka and Nogata2010). It has a high inter-specific variability along with low intra-specific variability (Hebert et al., Reference Hebert, Cywinska, Ball and deWaard2003a, Reference Hebert, Ratnasingham and deWaardb). In an environmental sample where a mixed population of species is present, a nested PCR approach has also been used to resolve the individual species (Patil et al., Reference Patil, Gunasekera, Deagle, Bax and Blackburn2005b). That study developed species-specific PCR assays for the detection of single species of a dinoflagellate, Gymnodinium catenatum in both environmental samples and in ballast water. The specificity of the primer showed that up to five cysts of G. catenatum can be detected in mixed populations. Similar results were achieved in detecting larval forms in Pacific oysters Crassostrea gigas (Patil et al., Reference Patil, Gunasekera, Deagle and Bax2005a), Tropical oyster C. belchiri (Klinbunga et al., Reference Klinbunga, Ampayup, Tassanakajon, Jarayabhand and Yoosukh2000) and in the sea star Asterias sp. (Deagle et al., Reference Deagle, Bax, Hewitt and Patil2003) using a PCR-based approach.
Fig. 2. PCR detection of Balanus amphitrite larvae in plankton sample. Left lane contains standard size markers (1 Kb DNA ladder, Chromous). Lane 1 is control. Lane 2 is zooplankton without cirrepede larvae. Lane 3 is PCR product of zooplankton. Lane 4 is zooplankton (without cirrepede larvae) spiked with 10 B. amphitrite larvae. Lane 5 is PCR product of zooplankton (without cirrepede larvae) spiked with 100 B. amphitrite larvae.
In the present study, efficiency of the PCR assay was enhanced by increasing the number of target species. Primer sensitivity was cross-checked with all available barnacle species in the study location. Balanus amphitrite specific primers did not amplify the other barnacle species, indicating that the primers were specific only to B. amphitrite. The presence of PCR inhibitors in the zooplankton sample was ruled out by conducting a PCR with zooplankton spiked with known numbers of B. amphitrite larvae. The resulting amplicons in these samples resulted in PCR success, ruling out the presence of any PCR inhibitors.
We demonstrate that the B. amphitrite larvae can be detected with extreme sensitivity by PCR amplification using the 18S species-specific primers designed in this study. Application of this method for detection of the B. amphitrite larvae in a mixed population can facilitate accurate screening of large numbers of samples and solve significant problems associated with larval ecology. This approach also can be used to differentiate B. amphitrite larvae from that of the closely related groups of barnacles within the mixed community of barnacles. Real-time PCR (qPCR) is recognized as an effective device for detection and quantification of different planktonic organisms in a mixed population. In future this tool can be adopted using the B. amphitrite specific primers designed in the present study for quantification of B. amphitrite in the plankton.
ACKNOWLEDGEMENTS
We are grateful to Dr Satish R. Shetye, the Director of the National Institute of Oceanography, Goa for his support and encouragement. We acknowledge the help provided by Dr Rakhee Khandeparker and other colleagues of BBD during this study. This work was carried out as a part of Ballast Water Management Programme, India that was funded by Directorate General of Shipping, Government of India. This is a CSIR-NIO contribution 5661.