



I. INTRODUCTION
Ferroelectric materials have attracted great interest in fundamental research owing to their potential technological applications (Du et al., Reference Du, Lei, Chen and Zhang2008; Gibbs et al., Reference Gibbs, Knight and Lightfoot2011; Malo and Maignan, Reference Malo and Maignan2012). YMnO3 exhibits multiferroicity with high ferroelectric and low antiferromagnetic transition temperature (Huang et al., Reference Huang, Cao, Sun, Xue and Chu1997; Lin et al., Reference Lin, Wang, Zhang, Wang, Zu, Song, Liu, Huang, Huang, Tao, Li, Bai, Li, Lei, Yu and Wu2019a, Reference Lin, Wang, Wu, Wang, Bai, Zu, Song, Wang, Liu, Li, Tao, Huang, Lei, Li and Wu2019b). It belongs to the non-center symmetric space group of P63cm at room temperature with typical ABO3-type structure (Prikockyte et al., Reference Prikockyte, Bilc, Hermet, Dubourdieu and Ghosez2011; Bi et al., Reference Bi, Wang, Hao, Lei, Dong and Zhou2019; He et al., Reference He, Luan, Wang, Wang, Du, Xu, Yang, Wang, Huang and Lei2019). A prototype ferroelectricity was observed in the so-called d0-ness systems, such as BaTiO3, in which Ti4+(d0) ions make off-center movements in TiO6 octahedra to lower the energy through enhanced Ti 3d and O 2p hybridization (Cohen, Reference Cohen1992; Ghosez et al., Reference Ghosez, Michenaud and Gonze1998; Babu et al., Reference Babu, He and Zhang2007). YMnO3 contains Mn (3d4) magnetic ions. Thus, a mechanism other than “d0-ness” is needed to account for the ferroelectricity. Possibilities include lone pair electrons as in BiMnO3 or a spin frustration of magnetic order as in TbMnO3 (Kimura et al., Reference Kimura, Goto, Shintani, Ishizaka, Arima and Tokura2003). But neither lone pair nor spin frustration can explain the ferroelectricity of YMnO3.
The ferroelectricity mechanism for YMnO3 has been ambiguous and confusing so far. Filippetti and Hill performed density functional theory (DFT) calculations and proposed that distortion was caused by hybridization between unoccupied Mn $3{\rm d}_z^2$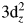 and O 2pz orbitals (Filippetti and Hill, Reference Filippetti and Hill2002). Cho performed the polarization-dependent X-ray absorption spectroscopy and concluded that Y 4d states were strongly hybridized with the O 2p states (Cho et al., Reference Cho, Kim, Park, Rho, Park, Noh and Kim2007). Liu proposed that the ferroelectric origin of YMnO3 was associated with the charge transfer from the Y–O bonds to the Mn–O bonds (Liu et al., Reference Liu, Huang and Qi2011), etc. However, these works did not systematically combine experiments with calculations when discussing the electronic structure of YMnO3.
and O 2pz orbitals (Filippetti and Hill, Reference Filippetti and Hill2002). Cho performed the polarization-dependent X-ray absorption spectroscopy and concluded that Y 4d states were strongly hybridized with the O 2p states (Cho et al., Reference Cho, Kim, Park, Rho, Park, Noh and Kim2007). Liu proposed that the ferroelectric origin of YMnO3 was associated with the charge transfer from the Y–O bonds to the Mn–O bonds (Liu et al., Reference Liu, Huang and Qi2011), etc. However, these works did not systematically combine experiments with calculations when discussing the electronic structure of YMnO3.
X-ray diffraction (XRD) and electron energy loss spectroscopy (EELS) are two important methods for characterization of materials and condensed matter. Combining with the XRD results, EELS can provide additional information. When incident electrons enter a material, they interact with the constituent atoms via electrostatic forces. As a result of these forces, some of the electrons are scattered. The interaction between incident electrons and the atomic electrons surrounding the nucleus is inelastic scattering. By analyzing the energy loss distribution of inelastic scattered electrons, the spatial environment information of the electrons can be obtained, and various physical and chemical properties of the sample can be studied. (Egerton et al., Reference Egerton, Crozier and Rice1987; Zhang et al., Reference Zhang, Yang, He, Wang and Li2008; Lin et al., Reference Lin, Wang, Zhang, Wang, Zu, Song, Liu, Huang, Huang, Tao, Li, Bai, Li, Lei, Yu and Wu2019a, Reference Lin, Wang, Wu, Wang, Bai, Zu, Song, Wang, Liu, Li, Tao, Huang, Lei, Li and Wu2019b; Wang et al., Reference Wang, Xu, Cai, Wang, Tao, Cui, He, Song and Zhang2019a, Reference Wang, Cui, Li, Lei, Li and Wei2019b). First-principle calculations were used to analyze the single-electron excitation from the valence band (VB) to the conduction band (CB) in EELS experiments. The empty density of states (DOS) can be comparable with the energy loss near-edge structure (ELNES) (Ikeno and Mizoguchi, Reference Ikeno and Mizoguchi2017; Wang et al., Reference Wang, Liu, Li, Lu, Wang, Zhao, Yuan, Cui, Li, Xin, Zhang, Lei and Lin2018). In this work, we combined the first-principle calculations with EELS experiments, valence EELS were obtained and analyzed by the calculated DOS, ELNES were investigated by comparing the simulated results with experiments to study the electronic structure of YMnO3. The results provided a theoretical basis for understanding the ferroelectric mechanism of YMnO3.
II. EXPERIMENTAL METHODS AND THEORETICAL DETAILS
The YMnO3 sample was prepared using a conventional solid-state reaction method (Zhang et al., Reference Zhang, Guo, Ge, Chen, Yao, Wang and Gu2014). The XRD measurements were performed in the EMPYREAN X-ray diffractometer from Dutch PANalytical company (with the detector PIXcel-3D). The powder sample was flattened onto the glass piece and loaded into the fixture. The measurements were carried out in the Bragg Brentano θ−2θ geometry with CuKα radiation at an operating voltage of 40 kV and an operating current of 40 mA. The samples were scanned from 10° to 130° with the step size of 0.26261°, and the dwell time is 39.53 s step−1. HighScore software was used to smooth the data, eliminate the alpha-2, and refine the lattice parameter. The EELS experiments were performed using a post-column Gatan Imaging Filter system attached to the microscope with an energy resolution of 1.0 eV for core-loss EELS. Its energy resolution was determined by the full-width half-maximum of the zero-loss peak. The spectra were acquired in selected area electron diffraction (SAED) mode at small momentum transfer. The energy dispersion is 0.2 eV pixel−1. All of the spectra were calibrated using the zero-loss peak position.
The calculations were performed using the DFT in Cambridge Serial Total Energy Package (CASTEP). Ultrasoft pseudopotential was expanded within a plane wave basis set to ensure the convergence with the cut-off energy (450 eV). Integrations in Brillouin zone were performed using special k-points generated with 4 × 4 × 2 mesh parameters grid. Exchange and correlation effects were described by Perdew–Burke–Ernzerhof (PBE) in generalized gradient approximation (Perdew et al., Reference Perdew, Burke and Ernzerhof1996). During the optimization, the convergence criteria of the energy and the maximum force were set at 1.0 × 10−5 eV atom−1 and 0.05 eV Å−1. The maximum stress was less than 0.1 GPa and the displacement of atoms convergence should be less than 0.002 Å.
III. RESULTS AND DISCUSSIONS
Figure 1 presents XRD patterns of the YMnO3 powders. Compared with the standard card (PDF 01-070-4962), the position of the diffraction peaks is almost the same, so it is shown that the powder sample is in P63cm hexagonal structure. Therefore, the YMnO3 of ferroelectric phase was determined by XRD experiments, which provided a qualitative basis for EELS experiments. And the standard card provides atomic structure information for the simulated calculation (a = 6.1390 Å, b = 6.1390 Å, c = 11.4070 Å, α = 90°, β = 90°, γ = 120°). Figure 2 presents the crystal structure of the optimized hexagonal YMnO3, the primitive unit cell contains six formula units (30 atoms). Two inequivalent Y1 (0, 0, 0.771) and Y2 (0.334, 0.667, 0.730) atoms form layers between the MnO5 triangular bipyramids tilted with respect to c-axis. Equivalent Mn atoms (0.335, 0.335, 0.497) are situated in the center of MnO5, which is surrounded by O3 (0, 0, 0.475), O41 (0.334, 0.667, 0.516), O42 (0.667, 0.334, 0.516) in the plane (OP) perpendicular to the c-axis, O1 (0.359, 0.359, 0.334) and O2 (0.308, 0.308, 0.660) along the c-axis (OT). The lattice constant and interatomic distance were obtained from the literature by Salazar-Kuri et al. (Reference Salazar-Kuri, Mendoza and Siqueiros2012), and we used these data to build the atomic structure of YMnO3. The related parameters and corresponding experimental values (Lima and Lalic, Reference Lima and Lalic2013) are presented in Table I. As it can be seen, the calculated parameters are consistent with the experimental data. The electron density distribution around the Mn ion in Figure 2(c) is anisotropic. The minimum electron densities of the Mn–OT bonds are larger than that of the Mn–OP bonds. The results can be described to the conventional localized bonding electrons, because the Mn–OT bonds (~1.9 Å) are quite shorter than the Mn–OP bonds (2.1 Å). Figure 3(a) is the SAED diagram under $[1\bar{1}0]$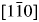 zone axis. Figure 3(b) shows the high-resolution transmission electron microscopy (HRTEM) results. The measured interplanar spacings were about 0.281 and 0.305 nm, which are comparable with the planes of (004) and (110) in the XRD, respectively. The matching planes shown by SAED are also (004) and (110). Moreover, the SAED pattern in Figure 2(a) is consistent with the FFT pattern [inset of Figure 2(b)] of the HRTEM image.
zone axis. Figure 3(b) shows the high-resolution transmission electron microscopy (HRTEM) results. The measured interplanar spacings were about 0.281 and 0.305 nm, which are comparable with the planes of (004) and (110) in the XRD, respectively. The matching planes shown by SAED are also (004) and (110). Moreover, the SAED pattern in Figure 2(a) is consistent with the FFT pattern [inset of Figure 2(b)] of the HRTEM image.

Figure 1. XRD pattern of the collected powders, the position of diffraction peaks is almost the same with the standard card (PDF 01-070-4962).

Figure 2. (a) Atomic structure of the P63cm ferroelectric YMnO3. (b) The triangular bipyramids of MnO5 in the atomic model. (c) Three-dimensional electron density distribution of YMnO3 obtained by CASTEP calculation. (d) Atomic structure with z-axis perpendicular to the paper surface.

Figure 3. (a) SAED pattern recorded from $[1\bar{1}0]$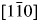 zone axis. (b) HRTEM image shows three different lattice directions, the left bottom shows the pattern after Fast Fourier Transform (FFT).
zone axis. (b) HRTEM image shows three different lattice directions, the left bottom shows the pattern after Fast Fourier Transform (FFT).
Table I. Calculated equilibrium lattice constants and selected interatomic distances (Å) in YMnO3 crystal compared to experimental data.

The single-scattering distribution S(E) of YMnO3 is shown in Figure 4(a), which is extracted by the removal of the plural scattering using Fourier-log deconvolution (Egerton, Reference Egerton2009). One important step to gain S(E) is the removal of the zero-loss peak, as emphasized by our previous work (Zhang et al., Reference Zhang, Qi, Jian and Duan2006), because it is high-energy tail covers features of the low-loss region (Rafferty et al., Reference Rafferty, Pennycook and Brown2000; Erni and Browning, Reference Erni and Browning2005). Above the bandgap, there are five well-resolved peaks, located at ~6.40 eV (A1), ~10.21 eV (A2), ~14.07 eV (A3), ~21.39 eV (A4) and ~34.49 eV (A5), respectively. The dominant peak A5 can be assigned to the bulk-plasmon oscillation. Other peaks originate from the single electron excitation from the VB to the empty DOS in the CB, and their profiles are expected to have a direct correlation with the joint DOS between occupied and unoccupied states in the energy bands. It should be noted, most of the peaks have mixed character since the dipole transitions selection rules have been extended. In the case of a large collection aperture, dipole-forbidden transitions (ΔL = 2) are sometimes observed (Egerton et al., Reference Egerton, Crozier and Rice1987; Lin et al., Reference Lin, Bai, Wang, Wang, Song, Huang, Wang, Wang, Li, Lei and Wu2017; Quhe et al., Reference Quhe, Liu, Wu, Yang, Wang, Li, Li, Yang, Peng, Lei and Lu2019). Thus, A1 is mainly attributed to the transitions between the O 2p to the Mn 3d band. A2 agrees with the characteristic of the O 2p to the Mn 3d/Y 4d transitions; meanwhile, the Mn 3d to the Y 4d transitions also make contributions to it. A3 corresponds to the excitation from the O 2p or Mn 3d to the Y 4d level. A4 corresponds to the transitions from the O 2s to the Mn 3d. For convenience, the assignments of the corresponding transitions were added to Table II. We found that every energy loss peak is related to the electronic transitions from the O 2p orbital to the Mn 3d or Y 4d orbitals. The information about electronic structure can be obtained from Figure 4(b). In the VB, the primarily populated O 2p and Mn 3d states form a block of the states whose energy lies approximately between −6.7 and −1.1 eV, within this energy range Mn atoms and O atoms exhibits hybridization. The energy range of −18.8 to −17.3 eV is mainly filled by a relatively narrow band of the 2s orbital electrons of O atoms. In the VB, only a small amount of Y 4d states are concentrated in the energy range from −6.6 to −1.7 eV, while the Y 4d states are mainly concentrated in the CB between 5.6 and 7.4 eV. The energy level from 2.8 to 7.1 eV is mainly occupied by the 3d orbital electrons of Mn atoms and is filled by little O 2p states. Overall, the local symmetry of the Mn is bipyramidal, thus the energy of its 3d orbital splits into doublets: in the VB top consists of the mixture of the e1g and a1g states, while the CB bottom is formed of the mixture of the a1g, e1g and t1g states (Sotero et al., Reference Sotero, Lima and Lalic2015). We also observed from the figure that the 4d orbital electrons of Y atoms and the 2p orbital electrons of O atoms have hybrid processes in both VB and CB, but the hybridization intensity is not stronger than Mn3d and O2p. Thus, the analysis of the DOS showed that the interaction among 2p of O, 3d of Mn, and 4d of Y atomic orbitals causes the spontaneous polarization, which results in the appearance of ferroelectric phase.

Figure 4. (a) The low energy loss spectrum of YMnO3. The intensity maximum around 34.49 eV is assigned to the bulk-plasmon loss, the smaller features are due to excitation from interband transitions. (b) Total and partial DOS in YMnO3. The fermi level is set at 0 eV.
Table II. Assignment of the major interband transition between experiment and theory.

To get more information about the electronic structure, we have carried out ELNES studies. The ELNES reflect both compositional analysis and degree of bonding hybridization. Thus, it contains valuable information about the nearest neighbored bonding (O 2p with the cationic d-orbital). Figure 5 shows the results of O K-edge by calculation and experiment. We can see that the simulated spectra nearly reproduce all details presented in the fine structure in terms of number of peaks, peak intensity and peak position. The O K-edge fine structure reflects mainly the density of O 2p states when hybridized with Y and Mn orbitals from the YMnO3. In Figure 5(b), five characteristic peaks marked by a, b, c, d and e can be clearly distinguished, where peaks a and b are related to the hybridization between O and Mn ions, and peaks c and d reflect the hybridization between O and Y ions. It indicates that O–Mn and O–Y covalent bonds are formed between O atoms and Mn/Y atoms, which has a stabilizing effect on the ferroelectric phase. The feature group of e is an absorption peak, which is a diffractive region due to a backscattering process between the absorber and its nearest neighbor oxygen shell (Kim et al., Reference Kim, Bhatnagar, Pippel, Alexe and Hesse2014; Wang et al., Reference Wang, Xu, Cai, Wang, Tao, Cui, He, Song and Zhang2019a, Reference Wang, Cui, Li, Lei, Li and Wei2019b). In Figure 6, there are two main peaks of Mn L2,3 edge in the experiment and calculations, which are originating from the electron transition from 2p3/2 and 2p1/2 states to unoccupied 3d bands (Nishida et al., Reference Nishida, Kobayashi, Kumamoto, Ikeno, Mizoguchi, Tanaka, Ikuhara and Yamamoto2013). The L3/L2 ratio is known to be related to the valence state of the 3d transition metal. And the ratio of Mn L3/L2 in this work was estimated using the method reported by Varela et al. (Reference Varela, Oxley, Luo, Tao, Watanabe, Lupini, Pantelides and Pennycook2009). The integrated areas of the Mn L2,3 edge used to estimate L3/L2 ratio are shown in Figure 6(c), and the ratio is about 2.46. It can be confirmed from related literature that the Mn of the YMnO3 compound is between trivalent and tetravalent oxidation states (Schmid and Mader, Reference Schmid and Mader2006). We simulated two valence states of Mn shown in Figures 6(a) and 6(b), and the simulated spectrum with Mn (+3) is in good agreement with the experimental spectrum. Therefore, we confirmed that the element of Mn is trivalent oxidation state in YMnO3.

Figure 5. (a) The simulation spectrum of O K-edge with core-hole effects. (b) The experimental spectrum of O K-edge.

Figure 6. (a) The simulation spectrum of Mn L2,3 edge in Mn (+4). (b) The simulation spectrum of Mn L2,3 edge in Mn (+3). (c) The experimental spectrum used to estimate the ratio of L3/L2. To extract the L3 and L2 peak intensities for the Mn L3/L2 ratio, a Hartree–Slater cross-section function was used as a step function scaled to the 10 eV wide region immediately to the right of the L2 peak. Then, the energy ranges of 10.0 eV of L3 and L2 peaks were integrated.
IV. CONCLUSIONS
In summary, the electronic structure of YMnO3 was investigated systematically based on both EELS and first-principle calculations. The XRD and SAED experiments revealed that the sample was pure hexagonal YMnO3 and the Mn is bipyramidal coordinated. Assignments of the individual interband transitions have been accomplished by comparing the interband transition energy with the calculated PDOS. The analysis from the DOS, low energy loss spectrum, and high-energy loss spectrum of the O K-edge, the ferroelectricity of YMnO3 is related to the hybridization (O 2p and Mn 3d, O 2p and Y 4d, respectively) and the forming of covalent bonds. In addition, the experimental spectrum of O K-edge is in good agreement with the simulated spectrum. The fine structure of Mn L2,3 edge confirmed that Mn ion in the YMnO3 belongs to trivalent oxidation state.
Acknowledgement We appreciate the useful suggestions from Xiaofeng Duan and Xing Lu. We also acknowledge the help of Zhiqiang Li during the cause of experiments.
FUNDING
This work was sponsored by National Natural Science Foundation of China under Grant Nos. 51672057, 51722205 and 51872034. This work was sponsored by Liaoning revitalization talents program under No. XLYC1807173 and the outstanding talents support the program by Dalian city under No. 2015R004. This work was also sponsored by Key Projects of Natural Science Foundation of Liaoning and Doctor Start-up Fund of Liaoning under Grant No. 20170520155.








