



Introduction
Trematodes of the family Paramphistomidae, known under the common name rumen fluke, are small pink pear-shaped concave amphistome flukes with two suckers, that infect a broad range of mammalian hosts including cattle, zebu, water buffalo, sheep, goats, deer and also llama and alpaca, and might have a significant impact on livestock productivity (Eduardo, Reference Eduardo1983; Deplazes et al., Reference Deplazes, Eckert, Mathis, von Samson-Himmelstjerna and Zahner2016; Mitchell et al., Reference Mitchell, Zadoks and Skuce2021). Even though adult rumen flukes are usually well tolerated (Taylor et al., Reference Taylor, Coop, Wall, Taylor, Coop and Wall2007), heavy infestation with the immature flukes can result in illness associated with the intestinal phase of the infection, usually termed acute larval paramphistomosis. It is a result of duodenal mucosa erosion (Taylor et al., Reference Taylor, Coop, Wall, Taylor, Coop and Wall2007; Millar et al., Reference Millar, Colloff and Scholes2012) and can manifest with dehydration, anaemia, hypoproteinaemia and submandibular oedema (Tilling, Reference Tilling2013). Severe cases are most prevalent in calves, while sheep and goats can exhibit clinical signs throughout their lives (Mason et al., Reference Mason, Stevenson, Cox, Dick and Rodger2012).
Paramphistomids have a complex life cycle involving aquatic or mud snails as intermediate hosts producing cercariae (Tilling, Reference Tilling2013; Horák et al., Reference Horák, Marchiondo, Colwell, Marchiondo, Cruthers and Fourie2019; Mitchell et al., Reference Mitchell, Zadoks and Skuce2021). Ruminants become infected by ingestion of metacercariae attached to grass while grazing or the floating metacercariae may be swallowed while drinking (Rondelaud et al., Reference Rondelaud, Vignoles, Vareille-Morel, Abrous, Mage, Mouzet and Dreyfuss2004). After excystation in the small intestine, young paramphistomes migrate ‘upstream’ and attach in the rumen, where they mature and lay eggs, which leave the host in feces (Fenemore et al., Reference Fenemore, Floyd and Mitchell2021).
Although rumen flukes are spread worldwide, paramphistomosis has been most common in temperate and tropical regions (mostly Asia, Africa and Australia), mainly in wet or flooded areas (Pfukenyi and Mukaratirwa, Reference Pfukenyi and Mukaratirwa2018). In Europe, paramphistomosis was historically regarded as being of minor importance. While the earlier studies from the middle of the 20th century report various Paramphistomum species, mostly Paramphistomum cervi and/or Paramphistomum leydeni in both cattle and game animals (Guilhon and Priouzeau, Reference Guilhon and Priouzeau1945; Willmott, Reference Willmott1950; Kotlán, Reference Kotlán1958; Deiana and Arru, Reference Deiana and Arru1963; Chroust, Reference Chroust1964; Kotrlá and Kotrlý, Reference Kotrlá and Kotrlý1982), more recent publications, especially from Western Europe, report predominantly Calicophoron daubneyi (Gordon et al., Reference Gordon, Roberts, Lean, Zadoks, Sargison and Skuce2013; Zintl et al., Reference Zintl, Garcia-Campos, Trudgett, Chryssafidis, Talavera-Arce, Fu, Egan, Lawlor, Negredo, Brennan, Hanna, De Waal and Mulcahy2014; Malrait et al., Reference Malrait, Verschave, Skuce, Van Loo, Vercruysse and Charlier2015; Huson et al., Reference Huson, Oliver and Robinson2017; Jones et al., Reference Jones, Brophy, Mitchell and Williams2017; Wenzel et al., Reference Wenzel, Küchler, Strube and Knubben-Schweizer2019; Bosco et al., Reference Bosco, Nocerino, Santaniello, Maurelli, Cringoli and Rinaldi2021). This parasite was originally described in the Kenyan highlands (Dinnik, Reference Dinnik1962); however, its known distribution range spans a large part of Europe and several African countries (Fig. 1). Calicophoron daubneyi infects mostly lymnaeid snails, typically Galba truncatula as its intermediate host, which allows the species to spread in the temperate climate (Sey, Reference Sey1979; Abrous et al., Reference Abrous, Rondelaud and Dreyfuss1996; Jones et al., Reference Jones, Williams, Dalesman and Brophy2015) and possibly outcompete the liver fluke Fasciola hepatica in the same host. Although the adults are thought to be well tolerated, cases of severe or even fatal disease outbreaks linked to heavy burdens by immature flukes have been reported across Europe (Mason et al., Reference Mason, Stevenson, Cox, Dick and Rodger2012; Millar et al., Reference Millar, Colloff and Scholes2012). With its increasing prevalence and negative impact on livestock production resulting in decreased milk yields and growth rates, C. daubneyi has been recognized as an emerging pathogen in Europe (Huson et al., Reference Huson, Oliver and Robinson2017).

Fig. 1. Distribution of Calicophoron daubneyi in Europe (A) and Africa (B). The parasite was first described in cattle from the Kenyan highlands (Dinnik, Reference Dinnik1962). Since then, C. daubneyi has been reported from cattle, zebu, water buffalo, sheep, moufflons, alpaca and llama across Africa and also Europe (red). However, DNA sequences are available only from a few countries (green). In the UK and Netherlands, the parasite's identity was confirmed by DNA (Ploeger et al., Reference Ploeger, Ankum, Moll, van Doorn, Mitchell, Skuce, Zadoks and Holzhauer2017; Mitchell et al., Reference Mitchell, Zadoks and Skuce2021); however, the sequences are not available. The map was created based on sequences published in GenBank and following literature: Dinnik (Reference Dinnik1962), Sey (Reference Sey1974, Reference Sey1978), Sey and Vishnyakov (Reference Sey and Vishnyakov1976), Sey and Arru (Reference Sey and Arru1977), Graber et al. (Reference Graber, Delavenay and Tesfamarian1978, Reference Graber, Chauve and Fonteneau1980), Kotrlá and Kotrlý (Reference Kotrlá and Kotrlý1982), Eduardo (Reference Eduardo1983), Abrous et al. (Reference Abrous, Rondelaud and Dreyfuss2000), Seck et al. (Reference Seck, Bâ and Marchand2008), Titi et al. (Reference Titi, Mekroud, Sedraoui, Vignoles and Rondelaud2010), Arias et al. (Reference Arias, Lomba, Dacal, Vázquez, Pedreira, Francisco, Piñeiro, Cazapal-Monteiro, Suárez, Díez-Baños, Morrondo, Sánchez-Andrade and Paz-Silva2011), González-Warleta et al. (Reference González-Warleta, Lladosa, Castro-Hermida, Martínez-Ibeas, Conesa, Munoz, López-Quílez, Manga-González and Mezo2013), Ferreras et al. (Reference Ferreras, Gonzalez-Lanza, Perez, Fuertes, Benavides, Mezo, González-Warleta, Giráldez, Martínez-Ibeas, Delgado, Fernándes and Manga-Gonzáles2014), Zintl et al. (Reference Zintl, Garcia-Campos, Trudgett, Chryssafidis, Talavera-Arce, Fu, Egan, Lawlor, Negredo, Brennan, Hanna, De Waal and Mulcahy2014), Malrait et al. (Reference Malrait, Verschave, Skuce, Van Loo, Vercruysse and Charlier2015), Sanna et al. (Reference Sanna, Varcasia, Serra, Salis, Sanabria, Pipia and Scala2016), Jones et al. (Reference Jones, Brophy, Mitchell and Williams2017), Ploeger et al. (Reference Ploeger, Ankum, Moll, van Doorn, Mitchell, Skuce, Zadoks and Holzhauer2017), O'Shaughnessy et al. (Reference O'Shaughnessy, Garcia-Campos, McAloon, Fagan, de Waal, McElroy, Casey, Good, Mulcahy, Fagan, Murphy and Zintl2018), Wenzel et al. (Reference Wenzel, Küchler, Strube and Knubben-Schweizer2019), Ates and Umur (Reference Ates and Umur2021), Bosco et al. (Reference Bosco, Nocerino, Santaniello, Maurelli, Cringoli and Rinaldi2021), Mitchell et al. (Reference Mitchell, Zadoks and Skuce2021), Padak and Karakus (Reference Padak and Karakus2021) and Wiedermann et al. (Reference Wiedermann, Harl, Fuehrer, Mayr, Schmid, Hinney and Rehbein2021). The maps were adjusted from https://www.mapsof.net/europe/europe-countries-map-blank?image=full and https://commons.wikimedia.org/wiki/File:BlankMap-Africa.svg.
The history of paramphistomosis in Czech Republic is comparable with the situation observed in Western Europe. Early studies from the 1950s and 1960s reported occasional occurrence of P. cervi in game animals (Erhardová and Kotrlý, Reference Erhardová and Kotrlý1955; Páv et al., Reference Páv, Zajíček and Zima1962) and acute paramphistomosis in young beef cattle (Chroust, Reference Chroust1964). Later, the spectrum was extended by Paramphistomum ichikawai and P. leydeni (Kotrlá and Chroust, Reference Kotrlá and Chroust1978; Kotrlá and Kotrlý, Reference Kotrlá and Kotrlý1982), however, the significance of paramphistomes was considered low as clinical signs and/or negative effect on milk/meat production were rarely observed. The latter study also reported occurrence of Calicophoron (formerly Paramphistomum) daubneyi in two moufflons from Eastern Bohemia (Kotrlá and Kotrlý, Reference Kotrlá and Kotrlý1982). At the turn of the 20th century, paramphistomosis was still considered of minor significance (Chroust, Reference Chroust and Teslík2000, Reference Chroust2006).
Identification of paramphistomid flukes by traditional morphology and morphometry is quite complicated and a thorough examination of histological sections is required (Eduardo, Reference Eduardo1982). On the contrary, DNA barcoding of the internal transcribed spacer 2 (ITS2) region represents a tool allowing identification to the species level also in the case of eggs in fecal samples as the source of the DNA (Mitchell et al., Reference Mitchell, Zadoks and Skuce2021). In this study, we aimed to investigate the prevalence and diversity of paramphistomid flukes in beef cattle herds across Czech Republic. In cooperation with beef farmers, we carried out large-scale coproscopic analyses to determine the prevalence of paramphistomid flukes and subsequently amplified the ITS2 region for species determination. The genetic diversity of the C. daubneyi population was evaluated using mitochondrial DNA (mtDNA) analysis.
Materials and methods
Fecal sample collection and coproscopy
The samples were collected by farmers using a sample collection kit provided within the project ‘Monitoring of strongylids involved in helminthiases of beef cattle in the Czech Republic and analysis of anthelmintic resistance and diversity’, implemented by the CEITEC VETUNI, University of Veterinary Sciences Brno (VETUNI), State Veterinary Institute Jihlava (SVI) and Czech Beef Cattle Association, and by the project team during visits to selected farms. Samples were collected as fresh as possible from the pastures, barns or directly from rectum during regular veterinary interventions in compliance with national ethical standards on animal handling. Samples were immediately transported to the project laboratories or sent by farmers using commercial parcel delivery services.
All fecal samples were processed by modified Sheather's sugar flotation (Sheather, Reference Sheather1923), simple sedimentation and Vajda's larvoscopy (Hovorka, Reference Hovorka1954). For the flotation, walnut size of feces was homogenized with water using a mortar and pestle, sieved to a tube and centrifuged at 2000 rpm for 3 min. The supernatant was discarded and a sugar solution of specific gravity 1.33 was added to the fecal sediment, vortexed and centrifuged again as described above. A surface film containing parasite stages was transferred onto a slide using inoculation loop, covered with a cover slip and examined under a light microscope. Approximately 3 g of feces were wrapped in a square of one layer gauze and put onto a small Petri dish with warm water (ca. 40°C). After 1 h, feces were removed, and the Petri dish was searched for emerged L1 larvae under a light microscope. Trematode eggs were detected by sedimentation, when the feces were mixed with 30 mL water, homogenized and sieved into a 50 mL beaker. After 12 min, the supernatant was decanted and the beaker with fecal sediment was filled again with water. This procedure was repeated every 10 min, until the supernatant was clean. After the last sedimentation cycle, the remaining 1–2 mL fecal sediment was poured into a small Petri dish and examined under a microscope to detect trematode eggs (Foreyt, Reference Foreyt2002; Deplazes et al., Reference Deplazes, Eckert, Mathis, von Samson-Himmelstjerna and Zahner2016). An Olympus BX41 microscope was used for examination of all samples. Photographs were taken on an Olympus BX53 equipped with a DP73 digital camera. Aliquots of ca. 5 g of selected fecal samples were stored at −20°C to create a biobank of material for subsequent DNA extraction.
Collection of adult rumen flukes
Adult paramphistomid flukes were collected from the rumen and reticulum of beef cattle at the abattoir during regular veterinary inspection. Protocol with the information about the host animal and the farm of origin was completed. Intensity of infection was classified using four categories:
-
+, sporadic occurrence of adult rumen flukes
-
++, 100–1000 adults
-
+++, 1000–10 000 adults
-
++++, mucosa (almost) continuously covered by adult flukes.
Several individuals of the rumen fluke were collected from each animal separately to a sterile vial and frozen at −20°C upon arrival to the laboratory.
DNA extraction
Selected fecal samples with observed paramphistomid eggs were used for the whole DNA extraction using the DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany) with the following modifications: (1) homogenizing of samples on the vortex was extended to 30 min, (2) incubation with C2 solution was extended to 30 min and (3) incubation with C3 solution was extended to be overnight. The DNA was eluted to 100 μL elution buffer included in the kit.
A NucleoSpin Tissue kit (Macherey-Nagel, Düren, Germany) was used for DNA extraction from individual adult paramphistomid flukes. Whole specimens were disrupted using a sterile micro-pestle and lysed in proteinase K overnight. In the final step, the DNA was eluted to 100 μL elution buffer.
DNA amplification
The second internal transcribed spacer region (ITS2) was amplified from both total fecal DNA and adult flukes. A total volume of 25 μL polymerase chain reaction (PCR) mixture contained 12.5 μL PCRBIO Taq mix red (PCR Biosystems Ltd., London, UK) and 1.25 μL 10 μ m each GA1 (5′-AGAACATCGACATCTTGAAC-3′) and BD2 (5′-TATGCTTAAATTCAGCGGGT-3′) primers (Luton et al., Reference Luton, Walker and Blair1992; Anderson and Barker, Reference Anderson and Barker1998; Lotfy et al., Reference Lotfy, Brant, Ashmawy, Devkota, Mkoji and Loker2010). The template DNA volume was 2 and 1 μL for the fecal DNA and adult helminth DNA, respectively, and 8 and 9 μL PCR-grade distilled water was used to complete the PCR mixture accordingly. Cycling conditions were as follows: initial denaturation 1 min at 95°C followed by 36 cycles of 15 s at 95°C denaturation, 15 s at 55°C annealing and 7 s at 72°C extension with final extension 2 min at 72°C.
A 855 bp fragment of mtDNA spanning 3′ end of cytochrome c oxidase subunit I gene (cox1), tRNA for threonine and 5′ end of mitochondrial rRNA large subunit (16S rRNA), which will be herein referred to as ‘mtDNA’, was amplified from the adult helminth DNA by primers Cd_Cox1F (5′-GCCGGGTCCTCAACATAATA-3′) and Cd_Cox1R (5′-AGCACAAAATCCTGATCTTACCA-3′) (Martínez-Ibeas et al., Reference Martínez-Ibeas, González-Warleta, Martínez-Valladares, Castro-Hermida, González-Lanza, Miñambres, Ferreras, Mezo and Manga-González2013). The same composition of the PCR reaction (25 μL) as for ITS2 amplification from the adults was used. Cycling conditions were as follows: initial denaturation 1 min at 95°C followed by 36 cycles of 15 s at 95°C denaturation, 15 s at 55°C annealing and 15 s at 72°C extension with final extension 5 min at 72°C.
All products were separated by electrophoresis in 1% agarose gel stained with Midori Green Advance (Nippon Genetics, Düren, Germany) and visualized on a UV transilluminator. PCR products were purified with ExoSAP-IT (Affymetrix, ThermoFisher Scientific, Czech Republic). If multiple bands were present, the bands of the expected size were cut from the gel and purified by using a PCR Cleanup kit (Geneaid Biotech Ltd., New Taipei, Taiwan). The products were sequenced commercially at Macrogen Europe (Amsterdam, Netherlands) using the amplification primers.
Sequence analyses
Sequences were checked, trimmed manually and assembled in Geneious Prime 2021.0.1 (http://www.geneious.com) and checked against BLAST (Altschul et al., Reference Altschul, Gish, Miller, Myers and Lipman1990). A median-joining network (Bandelt et al., Reference Bandelt, Forster and Röhl1999) was derived from mtDNA sequences using PopART (Leigh and Bryant, Reference Leigh and Bryant2015). Sequences of other paramphistomid trematodes were downloaded from GenBank, aligned with the obtained sequences using Clustal Omega implemented in Geneious Prime. Pairwise sequence distances were calculated from these alignments. The maximum-likelihood phylogenetic trees (ITS2, mtDNA) were calculated by IQ-TREE (Trifinopoulos et al., Reference Trifinopoulos, Nguyen, von Haeseler and Minh2016). The most suitable model was chosen by ModelFinder (Kalyaanamoorthy et al., Reference Kalyaanamoorthy, Minh, Wong, von Haeseler and Jermiin2017) implemented in IQ-TREE based on the highest Bayesian information criterion scores and weights. The tree topology was tested by 1000 replicates of ultrafast bootstrap (Minh et al., Reference Minh, Nguyen and von Haeseler2013), Shimodaira–Hasegawa (SH)-like approximate likelihood ratio test and approximate Bayes test (Anisimova et al., Reference Anisimova, Gil, Dufayard, Dessimoz and Gascuel2011).
Results
Paramphistomid flukes detected in most of Czech Republic regions
From March 2019 to June 2021, a total of 115 farms across the whole Czech Republic participated in the project and 5573 beef cattle fecal samples were sent to the laboratories of VETUNI and SVI. All but one of Czech Republic regions were represented between 1 and 26 farms per region. The number of samples per region varied from 34 in South Moravian Region (B) to 1299 in South Bohemian Region (C). Stages of at least one parasite were detected in 4752 samples (85.3%). The typical eggs of paramphistomid flukes (Fig. 2A and B) were identified in a total of 1668 fecal samples (29.9%); in 450 samples the paramphistomids were the only parasite detected. Paramphistomid flukes were detected in more than half of the participating farms (64%) in all but one region (S). The highest number of positive farms and samples was in the South Bohemian Region (Fig. 3). Percentage of positive samples varied among farms from 0.8 to 100%. In 24 farms (31.1%), more than 65% of the fecal samples were positive. These farms were in six regions: C (n = 8), K (n = 1), L (n = 2), P (n = 2), T (n = 7) and U (n = 3). In 25 farms (33.8%), paramphistomid eggs were detected in <20% samples. In the remaining 26 farms (35.1%), the percentage of positive samples was in the range of 21–60%. One to three samples from paramphistomid fluke-positive farms were selected for subsequent DNA barcoding.

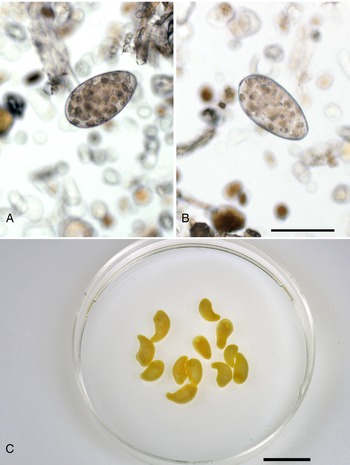
Fig. 2. Calicophoron daubneyi detected in Czech beef cattle herds. (A, B) Eggs detected by sedimentation technique, scale bar = 100 μm. (C) Adult flukes from rumen of a slaughtered cow from South Bohemia, preserved, scale bar = 1 cm.

Fig. 3. Percentage of fecal samples (green) and number of farms (in the parentheses) where paramphistomes were detected by coproscopy (left) out of the total number of farms participating (right) in the regions of Czech Republic. Regions A – Praha, B – South Moravian, C – South Bohemian, E – Pardubice, H – Hradec Králové, J – Vysočina, K – Karlovy Vary, L – Liberec, M – Olomouc, P – Plzeň, S – Central Bohemian, T – Moravian-Silesian, U – Ústí nad Labem, Z – Zlín. Adjusted from: CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=912955.
DNA barcoding showed all adult rumen flukes to be C. daubneyi
Adult rumen flukes (Fig. 2C) were collected from 90 animals originating from 78 farms in 11 regions (Table 1). The intensity of infection was evaluated in 56 host individuals. Most commonly (64%), light intensity of infection (++) was observed. In 12 animals, heavy infection with 1000–10 000 adult flukes was reported and one animal scored ++++ corresponding to rumen mucosa being (almost) completely covered by flukes.
Table 1. Overview of farms from which fecal samples and adult paramphistomid flukes were used for DNA barcoding with mtDNA haplotypes detected in adult rumen flukes

Farms where the fecal samples were sourced differed from farms where adult flukes originated from, only in South Bohemian Region (C) and Liberec Region (L), there was one farm where both fecal samples and adults originated from.
The ITS2 region (269–358 bp) was amplified in 88 C. daubneyi adult flukes originating from 77 farms in 11 regions. All sequences were identical. BLAST search showed 100% identity to two previously published sequences of C. daubneyi from Ireland (KP201674.1) and Italy (AY790883) and to nine C. daubneyi sequences directly submitted to GenBank originating from Turkey (MN044947.1), Algeria (LN610457-8.1) and South Africa (KT182091-96.1). The consensus sequence was submitted to GenBank under the accession number OL461703.
Ninety sequences (605–803 bp) of mtDNA were obtained from adult C. daubneyi specimens originating from 78 farms in 11 regions of Czech Republic (Fig. 3). In total, 13 individual haplotypes labelled CD1–CD13 and differing by 0.1–1.9% were recognized (Table 1). While haplotype CD13 was detected only in one region (C), haplotypes CD7 and CD12 were found in five districts. In the South Bohemian Region (C), 11 different haplotypes were detected (Table 1). Sequences were deposited to GenBank under accession numbers OL546359–OL546371.
A median-joining haplotype network was calculated from an alignment (717 bp) comprising 88 of our C. daubneyi mtDNA sequences and 22 C. daubneyi sequences downloaded from GenBank with 33 segregating sites, 16 parsimony-informative sites and nucleotide diversity (p = 0.0101). The haplotype network did not show any obvious clusters of certain haplotypes. Interestingly, almost all C. daubneyi haplotypes (n = 11) detected in the Czech beef cattle were previously reported in Ireland and one in Turkey (Fig. 4A). Only haplotypes CD6 and CD10 were recorded for the first time. The network comprised of six more unique haplotypes (CD14–CD19) and all originated in Ireland.

Fig. 4. (A) Median-joining haplotype network derived from 717 bp of C. daubneyi mtDNA. Haplotypes are indicated by code CD1–CD19. Origin of the sequence is indicated by the colour. Regions of Czech Republic are labelled by official abbreviations: C – South Bohemian, H – Hradec Králové, J – Vysočina, K – Karlovy Vary, L – Liberec, M – Olomouc, P – Plzeň, S – Central Bohemian, T – Moravian-Silesian, U – Ústí nad Labem, Z – Zlín, IRL – Ireland, TUR – Turkey. (B) Maximum-likelihood phylogenetic tree calculated by using the model TN + F + G4 from 731 bp of C. daubneyi mtDNA. Sequence of Calicophoron microbothrioides (GenBank accession number NC_027271.1) was used as an outgroup (not shown). Numbers at nodes are ultrafast bootstrap support/approximate Bayes test posterior probability/SH-like approximate likelihood ratio test, all in %. Only values above 75% are shown. Sequences derived in this study are indicated in bold blue and labelled by the haplotype code used in the haplotype network. Sequences downloaded from GenBank are labelled by the accession number. Haplotype code used in the network is indicated where appropriate.
A maximum-likelihood phylogenetic tree was calculated by using the model TN + F + G4 based on an alignment (731 bp) consisting of 13 of our C. daubneyi sequences, representing individual haplotypes CD1–CD13, 23 C. daubneyi sequences downloaded from GenBank and a Calicophoron microbothrioides (GenBank accession number NC_027271.1) sequence used as an outgroup. In the phylogenetic tree, a certain internal variability of analysed mtDNA was observed. A clade comprising Czech haplotypes CD1–CD4 and sequences corresponding to haplotypes CD16–CD19 distinguished in the haplotype network (Fig. 4) was formed and strongly supported by all three topology tests. However, the difference from other sequences varied from 0.2 to 2.2%.
Calicophoron daubneyi DNA detected in all examined fecal samples
The ITS2 region (279–339 bp) was amplified in 125 fecal samples originating from 49 farms in 12 regions (Fig. 3). Almost all sequences were identical, only two differed by one and two nucleotides, respectively. In all three polymorphic sites, a double peak A/C was observed at alignment position 156 in one sample and at positions 115 and 122 in the other sample. The ITS2 sequence obtained from the majority of the fecal samples matched the ITS2 sequence from adult rumen flukes and confirmed that all sequenced samples contained C. daubneyi DNA. All examined chromatograms were clear and there was no suspicion of mixed infection. Three sequences representing each detected variant were deposited to GenBank, with accession numbers OL461704, OL461706 and OL461707.
The final alignment (448 bp) of ITS2 region for a maximum-likelihood phylogenetic tree comprised of four sequences generated in this study representing adults of C. daubneyi and three sequences from fecal samples representing the individual variants, 172 sequences of paramphistomid trematodes downloaded from GenBank and two sequences of Ogmocotyle spp. as an outgroup. In the phylogenetic tree calculated by using the K2P + R2 model, all sequences of C. daubneyi formed one well-supported clade which was divided into two subclades. Our sequences clustered closely with sequences originating from Turkey, Algeria, South Africa, Italy and Ireland (Fig. 5). Most of the other clades in the tree corresponded to species, however, genera were mostly polyphyletic. Several clades (clades 1–7) comprised of sequences labelled as a few different taxa. For example, sequences labelled Cotylophoron cotylophorum and Calicophoron philerouxi appeared in each of two quite distant clades. Sequences labelled Calicophoron microbothrium clustered in four distinct clades. Clade 7 comprised of sequences labelled as five different taxa. The pairwise sequence distances were rather low across the whole tree, with 14.5% being the largest detected difference, but most of the sequences differed by <10%.

Fig. 5. Maximum-likelihood phylogenetic tree of the available paramphistomid trematodes' ITS2 region (448 bp) calculated by using the K2P + R2 model. Sequences derived in this study are indicated in bold blue, sequences downloaded from GenBank are labelled by the accession number and label provided in GenBank. For sequences of C. daubneyi, country of origin is indicated. Numbers at nodes are ultrafast bootstrap support value/approximate Bayes test posterior probability/SH-like approximate likelihood ratio test support value, all in %. Only values above 75 are shown. The branches were transformed to a proportional cladogram to illustrate the phylogenetic relationships better. The original tree without collapsed clades and true branch length pictured is provided as a Supplementary file S1. *ITS2 sequences from newly published study (Wiedermann et al., Reference Wiedermann, Harl, Fuehrer, Mayr, Schmid, Hinney and Rehbein2021) from Germany and Austria are identical with our consensus sequences but are not represented in the tree as the article was published after the analyses were completed.
Discussion
In this study, over 5500 fecal samples from 115 farms were collected and examined across the whole Czech Republic, representing possibly the most extensive study focusing on cattle parasites in the country so far. Paramphistomid flukes were detected in almost 30% of the samples, but at more than 64% farms which is in stark contrast to earlier studies of the livestock parasites in Czech Republic (Kváč, Reference Kváč2003; Chroust, Reference Chroust2006), but corresponds well with the trend observed across Western Europe (Szmidt-Adjidé et al., Reference Szmidt-Adjidé, Abrous, Adjidé, Dreyfuss, Lecompte, Cabaret and Rondelaud2000; Díaz et al., Reference Díaz, Paz-Silva, Sánchez-Andrade, Suárez, Pedreira, Arias, Díez-Baños and Morrondo2007; Zintl et al., Reference Zintl, Garcia-Campos, Trudgett, Chryssafidis, Talavera-Arce, Fu, Egan, Lawlor, Negredo, Brennan, Hanna, De Waal and Mulcahy2014; Malrait et al., Reference Malrait, Verschave, Skuce, Van Loo, Vercruysse and Charlier2015; Toolan et al., Reference Toolan, Mitchell, Searle, Sheehan, Skuce and Zadoks2015).
The identity of the detected paramphistomids was determined using 90 adult flukes and 125 fecal samples originating from a total of 125 different farms across the whole Czech Republic and the only detected species was C. daubneyi. It is not possible to predict that C. daubneyi is currently the only paramphistomid fluke infecting beef cattle in Czech Republic, but we can conclude that it is by far the dominant species. Kotrlá and Kotrlý (Reference Kotrlá and Kotrlý1982) examined over 400 rumen flukes collected from more than 3000 rumens of nine domestic and free-ranging ruminants and while the vast majority of the trematodes were identified as Paramphistomum spp., two moufflons hosted C. daubneyi representing the first record of the parasite in the country. It is possible that this parasite could spread from game animals to cattle over time. On the contrary, cattle have been a subject of an extensive international trade and thousands of animals have been annually imported to Czech Republic in the past few decades (Ministry of Agriculture of the Czech Republic, 2005, 2020). In 1995, Pavlásek (Reference Pavlásek1995) detected the number of parasite taxa, including Paramphistomum eggs in heifers imported to Czech Republic from Western Europe. Considering the broad distribution of C. daubneyi in Western Europe (Huson et al., Reference Huson, Oliver and Robinson2017), it is more probable that the parasite has been recently and probably repeatedly imported to Czech herds and consequently spread across the whole territory of Czech Republic.
The obvious lack of variability in the ITS2 is comparable to the situation observed for example in UK, Ireland, Netherlands, Germany, Austria and Italy (Rinaldi et al., Reference Rinaldi, Perugini, Capuano, Fenizia, Musella, Veneziano and Cringoli2005; Gordon et al., Reference Gordon, Roberts, Lean, Zadoks, Sargison and Skuce2013; Zintl et al., Reference Zintl, Garcia-Campos, Trudgett, Chryssafidis, Talavera-Arce, Fu, Egan, Lawlor, Negredo, Brennan, Hanna, De Waal and Mulcahy2014; Ploeger et al., Reference Ploeger, Ankum, Moll, van Doorn, Mitchell, Skuce, Zadoks and Holzhauer2017; May et al., Reference May, Brügemann, König and Strube2019; Wiedermann et al., Reference Wiedermann, Harl, Fuehrer, Mayr, Schmid, Hinney and Rehbein2021). On the contrary, 13 different haplotypes of mtDNA were detected across Czech Republic. In Ireland, Zintl et al. (Reference Zintl, Garcia-Campos, Trudgett, Chryssafidis, Talavera-Arce, Fu, Egan, Lawlor, Negredo, Brennan, Hanna, De Waal and Mulcahy2014) and O'Shaughnessy et al. (Reference O'Shaughnessy, Garcia-Campos, McAloon, Fagan, de Waal, McElroy, Casey, Good, Mulcahy, Fagan, Murphy and Zintl2018) found 16 and 20 different haplotypes, respectively. As C. daubneyi is currently regarded as an emerging pathogen, determination of the parasite origin could help predict its further spread. Only two of our 13 haplotypes were detected for the first time, suggesting common evolutionary history with the Irish C. daubneyi population and only a recent separation. The ITS2 has proven to be a good marker for screening and determination of paramphistomid trematodes as suggested also by Mitchell et al. (Reference Mitchell, Zadoks and Skuce2021). However, the ITS2 analysis revealed several discrepancies between the sequence annotation and its genetic identity (Fig. 5). Phylogenetic analyses of Paramphistomidae cox1 carried out in the study of Laidemitt et al. (Reference Laidemitt, Zawadzki, Brant, Mutuku, Mkoji and Loker2017) and newly published study of Wiedermann et al. (Reference Wiedermann, Harl, Fuehrer, Mayr, Schmid, Hinney and Rehbein2021) showed a slightly better resolution of the species-corresponding clades; however, some polytomies and clades with very low support were present. Additionally, few sequences, e.g. KX670107 Calicophoron phillerouxi clustered in a clade different from the sequence annotation, which was C. microbothrium in this case (Wiedermann et al., Reference Wiedermann, Harl, Fuehrer, Mayr, Schmid, Hinney and Rehbein2021). Laidemitt et al. (Reference Laidemitt, Zawadzki, Brant, Mutuku, Mkoji and Loker2017) observed discrepancy between nuclear (ITS2) and mitochondrial (cox1) identity in several paramphistomid specimens and suggested a possible hybrid origin of these individuals. However, observing a discrepancy between the sequence annotation and its genetic identity, misidentification of the specimens used for DNA extraction may be the reason. As some studies use stages from mollusc intermediate hosts and/or eggs, the morphological determination is limited (Laidemitt et al., Reference Laidemitt, Zawadzki, Brant, Mutuku, Mkoji and Loker2017). Morphology of adult paramphistomid flukes is also quite intricate (Eduardo, Reference Eduardo1982, Reference Eduardo1983); however, performing the accurate morphological identification of reference specimens before the DNA sequencing is crucial to enable easy DNA barcoding in the future studies.
The prevalence varied among the farms and in about a third of them, more than 65% of the examined fecal samples contained paramphistomid eggs. A shift in the spectrum of paramphistomids of cattle also has a range of ecological connotations. European species of the Paramphistomum genus typically exploit planorbid aquatic snails as their intermediate hosts. In contrast, the dominant intermediate host of C. daubneyi is G. truncatula (and a few other lymnaeid snails) (Huson et al., Reference Huson, Oliver and Robinson2017; Atcheson et al., Reference Atcheson, Skuce, Oliver, McNeilly and Robinson2020). Beef cattle production in Czech Republic has moved to areas with higher altitude with conditions perfect for G. truncatula, but not for most of the planorbids. Importantly, the same spectrum of molluscs is involved in the life cycle of the liver fluke F. hepatica (Abrous et al., Reference Abrous, Rondelaud and Dreyfuss2000; Deplazes et al., Reference Deplazes, Eckert, Mathis, von Samson-Himmelstjerna and Zahner2016; Beesley et al., Reference Beesley, Caminade, Charlier, Flynn, Hodgkinson, Martinez-Moreno, Martinez-Valladares, Perez, Rinaldi and Williams2018). Compared to C. daubneyi, F. hepatica is less prevalent in Czech Republic (Zmuda and Chroust, Reference Zmuda and Chroust2001) and also in the present study, the liver fluke was detected in fewer than 30 samples at 12 farms in five regions (data not shown). Fasciolosis had been causing significant economic loss in the past; however, the extensive use of anthelmintic treatment and pasture management have apparently reduced the liver fluke population in the last few decades of the 20th century. The herein reported common occurrence of C. daubneyi in the Czech farms indicates environmental conditions suitable also for F. hepatica and risk of its (re)emergence.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182021002158.
Data
All DNA sequences generated in this study were deposited to GenBank under accession numbers OL461703–OL461704, OL461706–OL461707 and OL546359–OL546371. Fecal samples and all DNA aliquots are deposited at the CEITEC VETUNI BioBank, University of Veterinary Sciences Brno, Brno, Czech Republic.
Acknowledgements
The authors thank all farmers involved in the study. They also thank the abattoirs for facilitating the collection of adult rumen flukes and are indebted to K. Špůrková and L. Hanáková for technical assistance in the lab. The authors are also indebted to Vincenzo Veneziano and Domenico Otranto as well as other colleagues from the parasitology teams of Università degli Studi di Napoli Federico II and Università degli Studi di Bari Aldo Moro for invaluable assistance and hints in sample processing. The authors thank the reviewers for their valuable comments that helped improving the manuscript.
Author contributions
B. Č., L. A., E. N., I. P., B. P., K. J., J. J. and P. P. participated in sample collection and coproscopic examinations. P. P. collected the adult rumen flukes and collected data on the infections. B. Č., L. A., E. N., K. J., J. J. and P. P. extracted the DNA. B. Č., L. A., E. N. and K. J. carried out DNA amplifications and processed samples for sequencing. L. A., E. N., K. J. and B. Č. processed the sequences. B. Č. and B. P. analysed the data from coproscopy. B. Č. analysed the sequences, carried out phylogenetic analyses, visualized the results and wrote the first version of the manuscript. D. M. and B. P. edited the manuscript. D. M., P. V. and K. M. secured the funding, coordinated and supervised the project. All authors read and agree with the manuscript.
Financial support
This study was supported by the Ministry of Agriculture of the Czech Republic (grant number QK1910204).
Conflict of interest
The authors declare none.
Ethical standards
All samples used in the study were collected in compliance with national ethical standards on animal handling and veterinary care.








{kind=link}