INTRODUCTION
Blood parasites of the phylum Apicomplexa and their avian hosts are valuable models to pursue important ecological, evolutionary, and behavioural questions (Atkinson and Van Riper III, 1991; Buchanan et al. 1999; Ricklefs et al. 2004; Sheuerlein and Ricklefs, 2004). Some studies found that these widespread parasites have negative effects on survival (Davidar and Morton, 1993; Sol et al. 2003), reproductive success (Merino et al. 2000; Marzal et al. 2005) and host body condition (Dawson and Bortolotti, 2000; Hatchwell et al. 2001), and thus may play a role in bird population dynamics. Also, haemosporidians in birds vary both over time and season, and between locations, in their apparent prevalence and parasitaemia which may lead to different selective pressures on bird populations (Allander and Bennett, 1994; Merilä et al. 1995; Valkiunias, 1997; Apanius et al. 2000; Sol et al. 2000; Bensch and Akesson, 2003).
Most of these studies determined prevalence based on microscopical examination of blood smears, and parasitaemia based on counts of parasites seen on these smears. However, these methods underestimate prevalence because the parasitaemia of many infections is too low to be detected visually. Molecular techniques (PCR amplification of a target gene segment) increase the efficiency of both parasite detection and identification (e.g. Tham et al. 1999; Richard et al. 2002; Fallon et al. 2003; Scopel et al. 2004; Waldenström et al. 2004; Beadell and Fleischer, 2005; Pérez-Tris et al. 2005). That is, some infections are missed during microscopical examination, but detected by use of a sensitive PCR-based assay. Microscopical study of smears and PCR can thus lead to contrasting results, especially if the data are used in correlative studies linking infection with some measure of host fitness. This problem is also important for ecological studies of prevalence that compare different sites or habitat types.
The more sensitive detection of infection of the PCR-based method also depends on the parasite species present because some species are less obvious on smears, perhaps because they more often remain at lower levels of parasitaemia (Waldenström et al. 2004). Thus, differences between results from microscopical studies and molecular detection appear to reflect variation in the typical parasitaemia per infected host (mean number of parasites in an infection). Standard PCR is not a quantitative method (infections are simply scored as infected or not infected). The question remains whether it is the parasitaemia of most infections or the prevalence of the parasite that is more important in understanding the impact of haemosporidians on host biology. Advances in quantitative molecular methods, especially real-time PCR (quantitative PCR) have only recently allowed quantitative screening of microparasites (e.g. Mouton et al. 2003; Matsuu et al. 2005; Contini et al. 2005).
The present study had 3 goals. First, a rapid molecular method using PRC followed by restriction Fragment Length Polymorphism (RLFP) analysis was developed (similar to the method of Beadell and Fleischer, 2005) to detect the specific parasites found in 2 populations of a single bird species, the European Blackbird Turdus merula. Thus, a sensitive test was developed to detect and identify parasites in infections. Second, a method was developed to obtain a reliable measure of parasitaemia within infections (including those infections not obvious under the microscope). To this end, a quantitative PCR method was used. Because one can expect a low power of detection when using blood smear examination in subpatent infection cases (weak infection), analysis of these cases by using q-PCR could act as a way to calibrate the measurements of low parasitaemia. Third, the 2 methods allowed comparison of prevalence and parasitaemia by host sex and between 2 sites (urban vs rural). The results using microscopical examination and the molecular methods were compared. Contrasted habitats were chosen because of the potential differences found in blood-sucking vectors (e.g. Grégoire et al. 2002; Sol et al. 2000), and host sex was analysed because a set of theoretical hypotheses predicted sex differences in parasitism distribution. Specifically, Hatchwell et al. (2000) have identified sex as a factor influencing Haemosporidian prevalence in European Blackbirds.
MATERIALS AND METHODS
Sampling
A long-term study of 2 European blackbird (Turdus merula) populations provided blood smears from a large sample of birds (n=289) (M. Barroca, unpublished results). These smears were examined to select 20 that represented the range of parasite morphology seen in infections (Haemoproteus and Plasmodium spp.). These infections were sequenced for a segment of the mitochondrial cytochrome b gene of the parasites (below). Then, to investigate parasite prevalence and parasitaemia, additional birds were sampled at 2 sites 35 km apart, an urban location (Dijon), and a forested habitat (Auxonne) (38 birds at each site). Blood was taken from each bird via the brachial vein to produce a thin blood smear to be dried, fixed in 100% methanol for 3 min, and stained in a Giemsa solution (5%, 45 min; Sigma) (Campbell and Dein, 1984). For molecular studies, 25 μl of blood was stored in 500 μl of lysis buffer (Seutin et al. 1991).
Parasite detection by blood smear examination
The smears were examined at 1000 magnification under oil using a Nikon E400 microscope. Parasites were identified to genus (Haemoproteus or Plasmodium). Parasites were counted within 10000 erythrocytes using a grid incorporated into the microscope eyepiece. If no parasites were seen, the bird was scored as not infected. As an estimation of parasitaemia, the number of parasitized cells was counted out of these 10000 erythrocytes, for each parasite type separately.
Molecular identification of blackbird haemosporidia parasites and prevalence screening by PCR-RFLP
DNA was extracted using a standard phenol-chloroform-isoamyl alcohol method (Sambrook et al. 1989). A negative control (containing no blood) was added to each extraction run to detect contamination. Purity and concentration of extracted DNA was determined with a spectrophotometer, and DNA diluted to a final concentration of 2 ng/μl.
Conserved PCR primers that would amplify a segment of the cytochrome b gene for all Haemosporidians (Plasmodium, Haemoproteus, and Leucocytozoon) were designed after alignment of 76 parasite sequences from diverse hosts (GenBank data). The alignment sequences are available at ftp.ebi.ac.uk, in directory pub/databases/embl/align, under Accession ALIGN_000970. We defined 2 new primers: HaemoFmod (5′ TTACAAGGTAGCTCTAATCCTTT 3′), which is the same as HaemoF (Perkins and Schall, 2002) but slightly shortened at the 3′ end, and CytbHaeR (5′ TTGTGGTAATTGACATCCAAT 3′). As a control of DNA quality, we also designed primers for the amplification of partial 18S rDNA specific of the host Turdus merula. They were designed after alignment of the 36 bird sequences (Alignment file available at ftp.ebi.ac.uk, under Accession ALIGN_000969). These primers were the following: OisF18 (5′ GTGAAACTGCGAATGGCTCAT 3′) and OisR18 (5′ CAAGATCCAACTACGAGCTTT 3′).
DNA was diluted 10-fold before amplification. PCR reactions were performed in a T3Thermocycler (Biometra), in 50 μl reaction mixtures adjusted to a final concentration of 1·3 mM MgCl2, 0·2 μM of each primer, 200 μM dNTPs and 1 U/μl of Eurobio Taq Polymerase with buffer according to the manufacturer's instructions. After initial denaturing at 95 °C, 35 cycles of 95 °C (1 min.), 50 °C (1 min.) and 72 °C (2 min.) were performed. The final extension was at 72 °C for 10 min. To control for contamination, we used a blank (no DNA added but water) every 10 reactions.
PCR products were sliced from the agarose gels, purified using an Agarose Purification Kit (Fermentas) and then sent to Macrogen for sequencing both strands. Sequencing was performed with the primers used for PCR. The sequences were submitted to the Blast server (http://www.ncbi.nih.gov/BLAST/Blast.cgi) for comparisons with known sequences. Then, a restriction map was produced for each sequence using Bioedit, to reveal specific endonuclease cutting sites. These enzymes were then used to digest PCR products, following the manufacturer's instructions (MBI Fermentas), allowing us to determine the prevalence of each parasite type in our samples.
Quantification of blackbird haemosporidian parasites by quantitative (real-time) PCR
Based on Cytochrome B sequence alignment of the three parasites (i.e. Haemoproteus TM, Plasmodium TM1, Plasmodium TM2), specific primer pairs were designed using the Primer3 program available online http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi (Table 1). These primer pairs allowed the amplification of a PCR fragment of about 200 bp of each of those parasites DNA. A fourth primer pair (Table 1) targeting the 18S rDNA of the host Turdus merula was also designed. It allows the estimation of 18S rDNA sequences of each DNA sample and was used as an internal standard to normalize q-PCR results.
Table 1. Primers designed for quantitative PCR (Parasite primers amplified cytochrome b, whereas host primers amplified 18S rDNA.)

Amplified fragments were cloned in Pgem-T Easy vector, following the recommendations of the manufacturer (Promega). Recombinant plasmids were purified using the QIAprep Spin Miniprep Kit (QIAGEN). Serial dilutions of appropriate cloned target sequence (from 0·2×108 to 0·2×102 copies) were used to elaborate calibration curves relating the log of the copy number (c) of the target sequence as a function of the Ct (cycle threshold) were developed, namely: log(c)=−3·3*Ct+36·01 (R2=0·99) for Haemoproteus TM, log(c)=−3·18*Ct+38·01 (R2=0·99) for Plasmodium TM1, log(c)=−3·2*Ct+33·66 (R2=0·99) for Plasmodium TM2, and log(c)=−3·2*Ct+30·95 (R2=0·99) for Turdus merula. For each run of quantitative PCR DNA blood samples and calibration standard samples were used as a template. Results were taken into account only when the coefficient of variation of the calibration curve was lower than 5%.
q-PCR reactions were carried out in a ABI 7900 (Applied Biosystems) using the following mix PCR preparation: 12·5 μl of Mix SYBRgreen PCR master mix including HotStar Taq™ DNA polymerase, Quanti Tec SYBR Green PCR buffer, dNTP mix with dUTP, SYBR Green I, Rox and 5 mM MgCl2 (QuantiTect™ SYBR® Green PCR kit, QIAGEN, France), 2·5 μl of each of the primer (5 pmol), 2·5 μl of H2O and 5 μl of DNA (2 ng/μl) for a final volume of 25 μl. Thermal cycling conditions were as follows: an initial cycle of 95 °C for 10 min, 8 cycles of 95 °C for 15 s, 60 °C for 30 s with a touchdown of −0·5 °C by cycle, 72 °C for 30 s, 40 cycles of 95 °C for 15 s and 56 °C for 30 s, 72 °C for 30 s and 1 cycle at 95 °C for 15 s, 56 °C for 15 s. Additional cycles of 56 °C for 15 s and 95 °C for 15 s were added to obtain dissociation curves specific from the targeted sequence, thereby allowing to check for the purity of the q-PCR products.
Parasite quantification per blood sample (i.e. parasitaemia) can only be relative to host blood cell quantity. For each individual sample, 2 reactions were performed, one targeting the 18S rDNA (host) and the other targeting cytochrome b (parasite). The relative parasitaemia of each sample was therefore given by the ratio: number of copies cytochrome b of parasites/number of copies of 18S rDNA of the host.
RESULTS
Molecular identification of blackbird Haemosporidia parasites
We found 3 unique sequences in the 20 surveyed infections. One sequence (GenBank , found in 7 infections) appears to be a species of Haemoproteus. BLAST search of GenBank data revealed that it is similar to other Haemoproteus (2 differences of 354 bases) (Perkins and Schall, 2002). The two other sequences (GenBank found in 10 infections, GenBank found in 3 infections) appear to be 2 species of Plasmodium (BLAST search). These 2 species differed by 8% of base positions, well within the genetic distance expected for different species of Plasmodium (Perkins and Schall, 2002; Ricklefs et al. 2004). We thus regard these as 3 parasite species (referred as Haemoproteus TM, and Plasmodium TM1 and TM2, respectively).
For each of the 3 sequences we obtained the restriction map on Bioedit and then choose an endonuclease which would enable us to discriminate the 3 different parasite genotypes. MunI was specific of the Haemoproteus strand, and BsrdI specific to Plasmodium TM2. SspI cut all 3 sequences but with different restriction patterns and was also used to give more confidence in our discrimination (SspI generated 2 fragments with the Haemoproteus sequence, 4 fragments with Plasmodium TM1, and 3 fragments with Plasmodium TM2).
Comparisons between detection methods
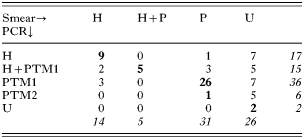
Compared to smear detection, the PCR-RFLP method improved blood parasite detection, since approximately 80% of individuals detected as negative were indeed infected by either Plasmodium or Haemoproteus (Table 2). This led to a significant difference in the proportion of uninfected individuals according to the detection method (34·21% vs 2·63% for smear and PCR methods, respectively, Fisher exact test two-tail, P<0·0001, n=76). The PCR-RFLP method also allowed detection of mistakes in parasite genus identification and detection of co-infections more efficiently than smear screening (Table 2). While smear methods provided only weak evidence that there was a deficit of co-infections (Fisher exact test, two tail: P=0·04), PCR analysis confidently confirmed that Haemoproteus – Plasmodium TM1 co-infections are more rare than expected by chance (Table 2, Fisher exact test, two tail: P<0·0001).
Table 2. The number of blackbirds infected by Haemoproteus parasites (H), Plasmodium parasites (P, PTM1 or PTM2, see text) and uninfected (U), as determined after smear screening (columns) and PCR-RFLP screening (rows) (Numbers in bold refer to animals where the infection status was detected by both methods; whereas normal numbers are those where there was inconsistency between smear and PCR methods (i.e. number of mistakes due to the low detection power of the smear method). Numbers in italics are totals of rows or columns.)
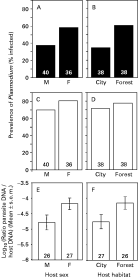
By analysing the effect of sex and habitat on Plasmodium prevalence using smear detection methods, a significant effect of both sex and habitat was found (logistic regression: whole model: χ23=11·00, P=0·01, effect of sex: χ21=5·47, P=0·02; effect of habitat: χ21=7·39, P=0·006; interaction sex*habitat: χ21=0·25, P=0·62; Fig. 1). However, the same analysis made with PCR data (to fit with the preceding analysis, the 2 Plasmodium species were grouped) revealed no significant effect of habitat, host sex and their interaction (Fig. 1; logistic regression: whole model: χ23=2·16, P=0·54). For Haemoproteus parasites, none of the similar analysis revealed a significant effect (logistic regression with smear data: whole model: χ23=0·41, P=0·94; logistic regression with PCR data: whole model: χ23=2·79, P=0·42; results not shown).

Fig. 1. Prevalence and intensity of haemosporidian parasites in Turdus merula. (A–D) Prevalence of Plasmodium parasites (Plasmodium TM1+Plasmodium TM2), revealed using different detection method (A and B) smear detection, (C and D) PCR-RFLP detection, as a function of host sex and host habitat. (E and F) Parasite DNA abundance of Haemoproteus and Plasmodium TM1 (relative to host DNA abundance) revealed by quantitative PCR, as a function of host sex and host habitat. Numbers in the bars are sample sizes. M are males, F are females. Differences in sample size between (A–D) and (E and F) are because abundance can accurately be estimated only in single infections.
Quantitative results as revealed by q-PCR
Despite our intention to design primer pairs specific for each parasite for q-PCR, cross-amplifications were seldom observed between Haemoproteus TM and Plasmodium TM1. Because of this lack of resolution, we were unable to obtain accurate quantitative results in co-infected individuals, and sample size was reduced to mono-infected individuals.
We controlled for whether the amount of parasite DNA (relative to host DNA) was correlated with the counts of parasites revealed by smears (i.e. where at least 1 parasite was counted). This analysis was impossible for Plasmodium TM2 and Haemoproteus, because of the small number of parasites detected on smears (n=1 and n=9, respectively). For Plasmodium TM1, the correlation was weak but significantly positive (rs=0·45, P=0·01, n=29).
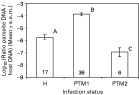
Parasitaemia estimated by q-PCR was different according to parasite species (Fig. 2, ANOVA on Log10 transformed data: F2,58=63·62, P<0·0001), with Plasmodium TM1 being significantly more abundant than others, and Plasmodium TM2 showing significantly lower parasitaemias than others (Fig. 2).

Fig. 2. Parasite DNA abundance in blackbirds (relative to host DNA abundance) revealed by quantitative PCR, as a function of parasite species. H: Haemoproteus; PTM1: Plasmodium TM1; PTM2: Plasmodium TM2. Numbers in the bars are sample sizes.
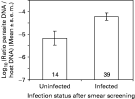
In Plasmodium TM1 and Haemoproteus, we investigated whether parasites were in lower numbers in ‘smear uninfected’ individuals (i.e. those where the infection was not detected on smears but detected by PCR-RFLP) than in ‘smear infected’ individuals (i.e. where the infection was detected by the 2 methods). The ANOVA included parasite species and smear infection status as factors, and their interaction. As seen before, Plasmodium TM1 was present in higher parasitaemia than Heamoproteus (F1,49=63·75, P<0·0001, Fig. 2). Parasites undetected by smear screening were in significantly lower number than those that were detected (F1,49=6·09, P=0·017, Fig. 3). There was no interaction between parasite species and smear infection status (F1,49=1·24, P=0·27) (Global model: F3,49=28·63, P<0·0001, r2=0·64). In addition, it should be noted that the parasitaemia was low for the Plasmodium TM2, for which most of the animals were not detected as infected by smear examination. The only individual where the infection was detected by smear had the higher parasitaemia (2·61 10−6 arbitrary units, compared to the 1·26 10−7 units obtained on average for this parasite in other samples).

Fig. 3. Parasite DNA abundance in blackbirds of Haemoproteus TM and Plasmodium TM1 revealed by quantitative PCR, as a function of infection status after smear screening. Animals declared as ‘uninfected’ after smear screening are in fact ‘false negative’ individuals. Numbers in the bars are sample sizes.
Finally, to check whether host factors might modulate parasitaemia, an ANOVA was made, including parasite species (Plasmodium TM1 and Haemoproteus TM), host sex (males vs females) and host habitat (city park vs forest) as factors, and their interactions. None of the interactions was significant (P>0·40), and they were therefore removed from the model. As already seen, parasite species influenced parasitaemia (F1,49=74·20, P<0·0001). In addition, females hosted significantly more parasites than males (F1,49=4·49, P=0·039, Fig. 1E), and blackbirds living in forest hosted significantly more parasites than those living in the urban park (F1,49=11·83, P=0·001, Fig. 1F) (Global model: F3,49=34·15, P<0·0001, r2=0·68). This phenomenon was not only due to a differential distribution of the weak infections (i.e. those not detected by smear screening, but detected by PCR), as their proportion did not significantly differ between host sex (P=0·54, D.F.=53) or between host habitat (P=0·12, D.F.=53), although a tendency exists for a higher proportion of weak infections in the urban habitat.
DISCUSSION
Turdus merula was infected by 3 genetically distinct strains of Haemosporidian in our study sites. Two were unambiguously close to avian Plasmodium identified previously (Perkins and Schall, 2002; Ricklefs et al. 2004; Szymanski and Lovette, 2005); and one close to avian Haemoproteus (Perkins and Schall, 2002; Ricklefs and Fallon, 2002). The high haemosporidian prevalence found here is of the same order as that estimated by Hatchwell et al. (2000) for a rural blackbird population in England. However, Hatchwell et al. (2000) observed Leucocytozoon more frequently than other genera, whereas this parasite was not detected in our study. Differences between these two studies in the haemosporidian community may reflect large-scale geographical variation (Scheuerlein and Ricklefs, 2004).
As previously found in various studies on bird malaria, the PCR-based method developed here allowed a better detection of parasites compared to smear screening (e.g. Richard et al. 2002; Waldenström et al. 2004). As a consequence, differences in prevalence attributed to host sex or habitat, as estimated by smear analysis, disappear after PCR-RFLP screening, mainly because almost all hosts were shown to be infected by one or the other haemosporidian parasite with the last method. Consequently, results on haemosporidian prevalence based on smear screening should be interpreted with caution, as suggested by Richard et al. (2002).
Quantitative PCR (qPCR) showed that the blood samples detected as negative in smears but positive in PCR-RFLP screening contained less parasite DNA than samples detected as positive using both methods. We therefore confirmed that lack of detection with smear screening was due to fewer parasites in blood samples. This result is in accordance with those obtained from a dilution experiment conducted by Fallon et al. (2003) and showing that detectability using PCR, decreased with parasitaemia. This observation led us to turn our attention from an analysis of prevalence to an emphasis of parasitaemia.
An interesting finding emerging from qPCR analysis is that there was significant variation in parasite numbers between parasite species. For instance, the parasitaemia for Plasmodium TM2 was 100–1000 weaker than that for Plasmodium TM1. The proximate reason for this difference may depend on several factors. First, parasite virulence and/or host capacity to limit the infection could vary between parasite strains, as interaction between parasite and host traits could explain variability in parasitaemia (Mackinnon and Read, 2004). Second, the abundance of respective vectors may differ in the field, allowing an increased transmission rate of one parasite relative to others (Sol et al. 2000; Scheuerlein and Ricklefs, 2004). Finally, the degree of parasite ‘sharing’ between blackbirds and other coexisting bird species may differ between parasite species and explain variation in parasitaemia (Bensch and Akesson, 2003).
Variations in parasitaemia were also found for individual parasite species. Here, Plasmodium TM1 and Haemoproteus TM parasitaemias were weaker in male hosts, and in urban habitats. Differences between the sexes in blackbirds may be explained by sex-specific immunity, or, alternatively, by sex-specific behaviour resulting in differences in exposure (Zuk and McKean, 1996; Tschirren et al. 2003). Differences between urban and forest habitats may be explained by differences in vector presence or/and abundance (Sol et al. 2003, Grégoire et al. 2002). Inter-population variation in host resistance against avian malaria may also explain such a result, as revealed by recent studies on MHC allele diversity in natural populations of house sparrows Passer domesticus (Bonneaud et al. 2006). As our study was not designed to test these hypotheses, future studies, using q-PCR, focusing for example on parasite dynamics within host individuals, should be able to address these questions more clearly.
The general conclusion of this study is that differences in parasitaemia, as estimated with qPCR, were comparable to differences in parasite prevalence estimated by smear screening. Standard PCR methods (like our PCR-RFLP procedure) therefore obscure patterns detected by smear or qPCR. This suggests that previous results obtained by smear screening (e.g. Buchanan et al. 1999; Dawson and Bortolotti, 2000; Hatchwell et al. 2001; Sol et al. 2003) may be re-interpreted in terms of parasitaemia instead of parasite prevalence.
We thank Gabriele Sorci for comments and discussions, and Mike Cherry for reading the manuscript. M. Barroca's thesis was funded by a grant from the Ministère de l'Education Nationale et de la Recherche. A part of this study was funded by the Institut Français de la Biodiversité, programme ‘Origine, distribution et dynamique de la biodiversité’. We thank the CRBPO for capture authorization and the Ville de Dijon for open access to urban parks.