


INTRODUCTION
Red foxes (Vulpes vulpes) play an important role in the transmission of a broad spectrum of pathogens of medical and veterinary importance, and this is mostly due to their increasing population density, wide distribution, high susceptibility to relevant pathogens and their good adaptation to urban and periurban environments (Duscher et al. Reference Duscher, Leschnik, Fuehrer and Joachim2015). Increasing number and urbanization of foxes consequently increase the risk of pathogen transmission to humans and domestic animals (Barandika et al. Reference Barandika, Espí, Oporto, Del Cerro, Barral, Povedano, García-Pérez and Hurtato2016). In the last few years, foxes have been intensively investigated worldwide for the presence of protozoan (e.g. Babesia, Hepatozoon, Leishmania), bacterial (e.g. Anaplasmataceae, Bartonella, Borrelia) and filarial (e.g. Dirofilaria) infections transmitted by haematophagous arthropod vectors, as they seem to play an important role in the maintenance and transmission of the agents (Henn et al. Reference Henn, Chomel, Boulouis, Kasten, Murray, Bar-Gal, King, Courreau and Baneth2009; Torina et al. Reference Torina, Blanda, Antoci, Scimeca, D'Agostino, Scariano, Piazza, Galluzzo, Giudice and Caracappa2013; Millán et al. Reference Millán, Ferroglio and Solano-Gallego2014; Penezić et al. Reference Penezić, Selaković, Pavlović and Ćirović2014; Cardoso et al. Reference Cardoso, Gilad, Cortes, Nachum-Biala, Lopes, Vila-Viçosa, Simões, Rodrigues and Baneth2015; Dumitrache et al. Reference Dumitrache, Matei, Ionică, Kalmár, D'Amico, Sikó-Barabási, Ionescu, Gherman and Mihalca2015; Hodžić et al. Reference Hodžić, Alić, Fuehrer, Harl, Wille-Piazzai and Duscher2015). The incidence, diversity and geographic range of the pathogens harboured by red foxes and other wild carnivore species have increased in the recent past due to several factors, and these mainly include climate changes, destruction of wildlife habitats, urbanization, but also increased awareness of the scientific community and establishment of more reliable and accurate diagnostic tools (Aguirre, Reference Aguirre2009; Alvarado-Rybak et al. Reference Alvarado-Rybak, Solano-Gallego and Millán2016).
However, many aspects of the epidemiology of vector-borne diseases such as host range, vector host use, conservation threats, possible non-vectorial routes of transmission and zoonotic potential are still poorly understood. Therefore, investigation of the pathogen distribution, vector-host–pathogen associations and dynamics of infections among wild and domestic carnivores are of a great importance for better understanding the epidemiology and also for filling the existing gaps required for establishing efficient control strategies (Otranto et al. Reference Otranto, Cantacessi, Pfeffer, Dantas-Torres, Brianti, Deplazes, Genchi, Guberti and Capelli2015; Alvarado-Rybak et al. Reference Alvarado-Rybak, Solano-Gallego and Millán2016).
The data on the occurrence and diversity of vector-borne pathogens in foxes in Austria are fragmentary and limited to a very small area located in the north-eastern part of the country (Gänserndorf, Lower Austria) (Duscher et al. Reference Duscher, Fuehrer and Kübber-Heiss2014). Therefore, this comprehensive molecular study focused on the following aspects: (i) prevalence and genetic diversity of pathogens transmitted by hematophagous arthropods in a red fox population in western Austria; (ii) genetic variations and haplotype composition of the two most common apicomplexan haemoparasites in foxes namely Babesia cf. microti and Hepatozoon canis; (iii) comparative suitability of blood and spleen tissues for molecular detection of haematozoa, and (iv) possible vertical transmission of H. canis from an infected vixen to its offspring, and importance of the non-vectorial transmission pathway in the circulation of this parasite.
MATERIAL AND METHODS
Study area and sample collection
The study was conducted in the two westernmost Austrian provinces, Tyrol and Vorarlberg between November 2013 and March 2015. Carcasses of 506 red foxes (245 males and 261 females) were collected as a part of Trichinella and Echinococcus multilocularis control programs, and delivered to the Institute for Veterinary Disease Control in Innsbruck, Austria. A complete necropsy was performed and spleen tissue was sampled from all animals and stored at −20 °C until processed for molecular analysis. In addition, blood samples from 351 foxes were taken from body cavities (primarily thoracic cavity) and spotted (100 µL) onto filter paper. Animals were sexed and divided into two age groups based on tooth wear: juveniles (<1 year; 337 individuals) and adults (⩾1 year; 169 individuals).
DNA extraction, PCR amplification and sequencing
Genomic DNA was isolated from the spleen tissue with DNeasy® Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. DNA extraction from the blood spots was performed using the protocol previously described (Fuehrer et al. Reference Fuehrer, Starzengruber, Swoboda, Ali Khan, Matt, Ley, Thriemer, Haque, Yunus, Hossain, Walochnik and Noedl2010). Briefly, disks of spotted and air dried blood, 4 × 4 mm2 in size, were cut out of the filter paper and soaked in 100 µL phosphate-buffered saline (PBS) at 4 °C overnight. After the isolation step, DNA was purified with InstaGene™ Matrix (Bio-Rad, Austria). DNA extracts from spleen and blood samples were used as templates for polymerase chain reaction (PCR) amplification and molecular detection of pathogens. For subtyping of genetic variants, all samples positive for Anaplasma phagocytophilum in the 16S rRNA gene were additionally tested by PCR targeting the groEL heat shock operon segment. Primer sequences, targeted genes and protocols used in the study are listed in Table 1. All PCR reactions were carried out in a final volume of 25 µL with 5× Green Reaction Buffer and GoTaq G2® Polymerase (Promega, Germany). Positive (specific pathogen DNA i.e. B. canis, H. canis, L. infantum, A. phagocytophilum, E. canis, B. henselae, R. raoultii, D. repens, D. immitis) and negative (PCR grade water) controls were included in PCR assays. The amplified products were electrophoresed on 2% agarose gel stained with Midori Green Advance DNA stain (Nippon Genetics Europe, Germany), and purified and sequenced in both directions (Sanger sequencing method) by a company LGC Genomics, Germany. The sequences obtained were edited with the software BioEdit v.7.2.5 (Hall, Reference Hall1999) and compared for identity with the sequences deposited in GenBank® nucleotide database using BLAST (Basic Local Alignment Search Tool) analysis (http://www.ncbi.nlm.nih.gov/BLAST).
Table 1. Primer sequences and PCR protocols used for molecular detection of arthropod-borne pathogens

Molecular detection of Hepatozoon canis in foetal tissues
A total of six foetuses from a pregnant fox that was found to be infected by H. canis (universal primers) were dissected and tested by PCR in order to investigate the possible transplacental infection. Briefly, the partially developed foetuses (5–6 cm in length) were cut independently in two halves by means of a sterile scalpel and all internal organs from abdominal and thoracic cavity were removed, transferred to a 50 mL plastic tube and then homogenized using a metal spatula. Standard precautions (e.g. separate gloves, sterile dissection tools, sterile tubes) were taken during dissection to avoid contamination and contact with infected blood from the vixen. Approximately 20 mg of foetal tissue homogenate was subjected to DNA extraction using the same kit (DNeasy® Blood & Tissue Kit, Qiagen). The DNA from the mother fox and all six foetuses were tested with genus-specific primers H14Hepa18SFw and H14Hepa18SRv (Hodžić et al. Reference Hodžić, Alić, Fuehrer, Harl, Wille-Piazzai and Duscher2015), which amplify a 620 bp fragment of the 18S rRNA gene of Hepatozoon species (Table 1), and positive products were sequenced.
Phylogenetic analysis
For phylogenetic analysis, only those 18S rRNA nucleotide sequences of Babesia cf. microti and H. canis available in GenBank® database, which showed a minimum of 458 bp (86 sequences) and 413 bp (370 sequences) overlap respectively, with sequences generated herein were included in the overall alignment. Multiple sequence alignment was performed with ClustalW algorithm implemented in BioEdit v.7.2.5 (Hall, Reference Hall1999) and the sequences were trimmed manually. Median Joining (MJ) networks from both datasets were calculated with Network software v.4.6.0.0 (available at: www.fluxus-engineering.com) applying the default parameters. Unnecessary median vectors were reduced in the post-processing step using Maximum parsimony (MP) option.
Statistical analysis
The Mann–Whitney-U test was employed to test the associations between pathogen occurrence and sex and age of the animals, whereas proportions of positivity between blood and spleen were compared with the Chi-Squared and Fisher's exact tests. Differences were considered statistically significant if P < 0·05. All statistical analyses were performed with SPSS 20.0 statistical software. Samples positive for Babesia canis, A. phagocytophilum, Candidatus Neoehrlichia sp. and Bartonella rochalimae were excluded from the statistical analysis because of very low infection rates.
Ethical statement
All animals from the present study were killed legally during regular hunting events and under the Austrian hunting laws.
RESULTS
Pathogen diversity, prevalence and multiple infections
Overall 69·5% blood samples (244/351; 95% CI: 64·5–74·1%) and 56·7% spleen tissue samples (287/506; 95% CI: 52·4–60·1%) of red foxes from western Austria were found to be positive by PCRs and the following pathogens were identified: B. canis, Babesia cf. microti (syn. Theileria annae), H. canis, A. phagocytophilum, Candidatus Neoehrlichia sp. and B. rochalimae (Table 2). Multiple infections with two or three different agents were recorded in 33 (6·5%) and 1 (0·2%) animals, respectively, and co-infection with Babesia cf. microti and H. canis was most prevalent (31/506; 6·1%) followed by H. canis and A. phagocytophilum (2/506; 0·4%), and Babesia cf. microti, H. canis and A. phagocytophilum (1/506; 0·2%). DNA of Babesia cf. microti and H. canis was detected in both blood (50·7 and 18·5%, respectively) and spleen tissue (25·7 and 29·8%, respectively), whereas A. phagocytophylum (0·6%), Candidatus Neoehrlichia sp. (0·4%) and B. rochalimae (0·2%) could be amplified only in spleen samples. Babesia canis DNA from a single positive fox (0·3%) was found in blood, but not in spleen (Table 2). Furthermore, blood was significantly more frequently infected with Babesia cf. microti (P < 0·0001) compared with spleen, but the positivity rate of H. canis infection was higher in spleen (P < 0·05) than in blood (Table 2). However, no significant differences were observed between infection rates and gender or age (P > 0·05) of the host.
Table 2. Prevalence of pathogens detected in blood and spleen samples of red foxes (Vulpes vulpes) in western Austria

*P < 0·05; ***P < 0·0001.
Sequence and phylogenetic analyses
A total of 244 blood and 281 spleen samples were positive by PCR performed with universal primer sets BTH-1R, BTH-1R and GF2, GR2, which amplify the 18S rRNA gene of piroplasmids and Hepatozoon species (Tables 1 and 2). However, only 378 (178 blood and 200 spleen) samples, which delivered a strong signal were sequenced, and the BLAST search revealed 99–100% identity to the following nucleotide sequences: B. canis (100%, e.g. GenBank® accession nos. KY447296, KT008057), Babesia cf. microti (99–100%, e.g. KY447297, KY486299, KT223483) and H. canis (100%, e.g. KU893127, FJ497011). The species differentiation from the remaining PCR products (66 blood and 81 spleen samples) was made based on the fragment length, as there is an approximately 30–40 bp difference between Babesia and Hepatozoon species (Table 1) (Modrý et al. Reference Modrý, Beck, Hrazdilová and Baneth2017).
Three samples positive by Anaplasmataceae-specific PCR showed 100% identity to the 16S rRNA gene of A. phagocytophilum strains (e.g. KY458571, KX180948), whereas two nucleotide sequences belonged to the recently described sequence of Candidatus Neoehrlichia sp. [the finding of one of these samples has already been published in Hodžić et al. (Reference Hodžić, Cézanne, Duscher, Harl, Glawischnig and Fuehrer2015a )]. Anaplasma phagocytophilum species validation and subtyping were done by PCR targeting groEL gene and sequence analysis revealed 100% identity to a genetic variant (so called G-variant, A → G) previously detected in humans (e.g. KT454993, KF015601), and domestic and wild animals (e.g. CP006618, JF494840, EU860090, DQ680012, KR092132). Furthermore, the spleen sample from one fox was infected by a zoonotic bacterium, B. rochalimae (99% identity to e.g. DQ683199, KX169194). None of the collected samples showed the presence of Ehrlichia, Leishmania, Rickettsia, Dirofilaria and other filarioid helminths DNA.
The MJ network constructed with 18S rRNA sequences of Babesia cf. microti obtained in this study and sequences available in GenBank® showed the existence of ten different haplotypes separated from one another by one to three nucleotide substitutions (Fig. 1). Almost all the sequences from our study, except one with a single nucleotide position difference, belong to the most dominant and centrally positioned haplotype, along with sequences previously reported in domestic and wild carnivores (Fig. 1A) from Europe, Asia and North America (Fig. 1B). The genealogic network of H. canis showed a considerable 18S rRNA gene nucleotide diversity compared to Babesia cf. microti, with a total of 56 individual haplotypes divided into several clades and with no obvious correlation between geographic locality, host species and haplotype composition (Figs 2 and 3). However, only one haplotype which grouped together with a haplotype described in other carnivores originating from several European countries was found circulating in the fox population in western Austria (Figs 2 and 3).
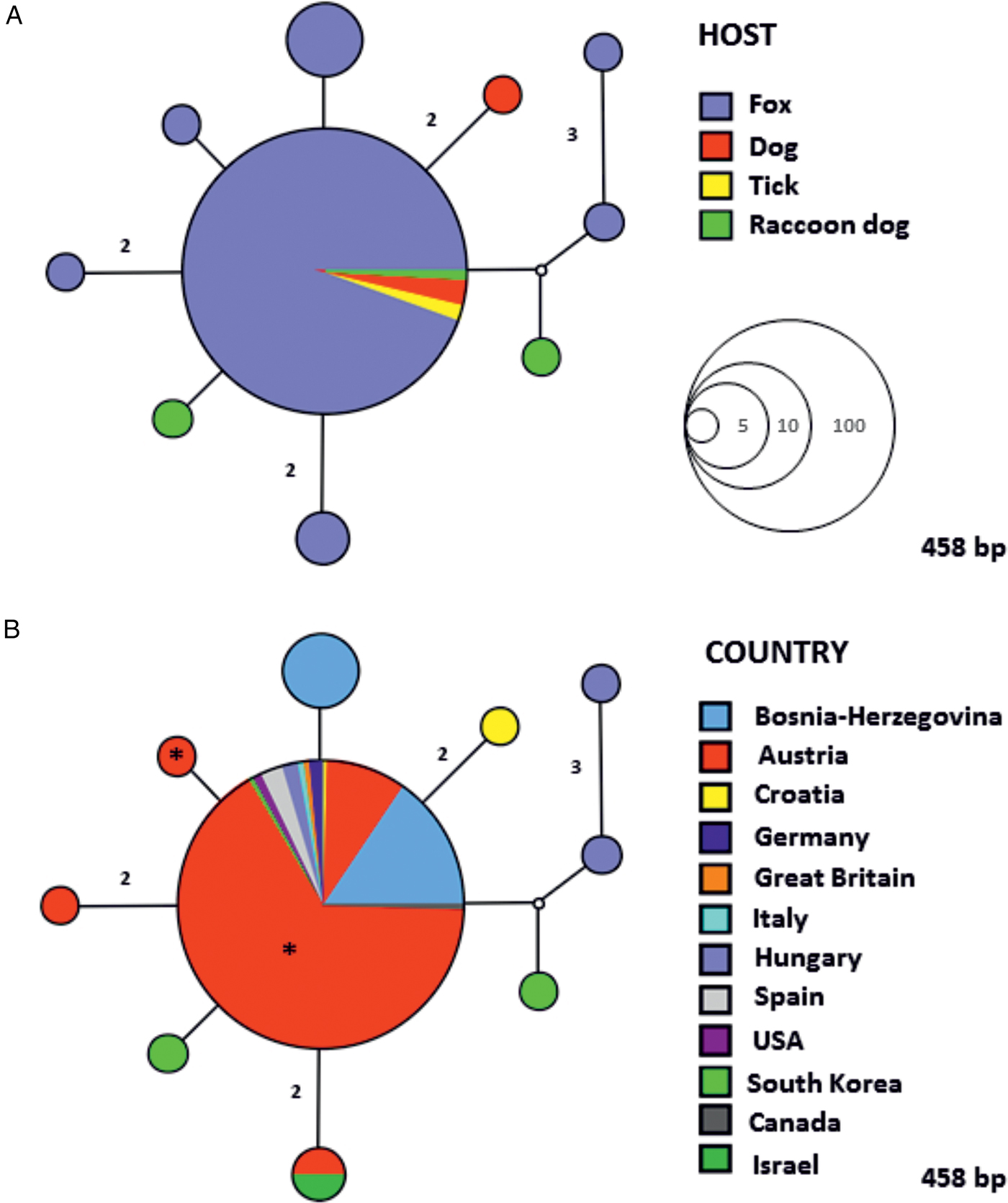
Fig. 1. Median Joining (MJ) network of the 18S rRNA gene (458 bp) of Babesia cf. microti, showing the relationships between haplotypes according to host (A) and geographic locality (B). The size of the circles in the network is proportional to the number of individuals sharing the same haplotype. Numbers along branches indicate the number of substitutions between haplotypes. An asterisk (*) in the circles represents sequences (haplotypes) obtained in the present study.
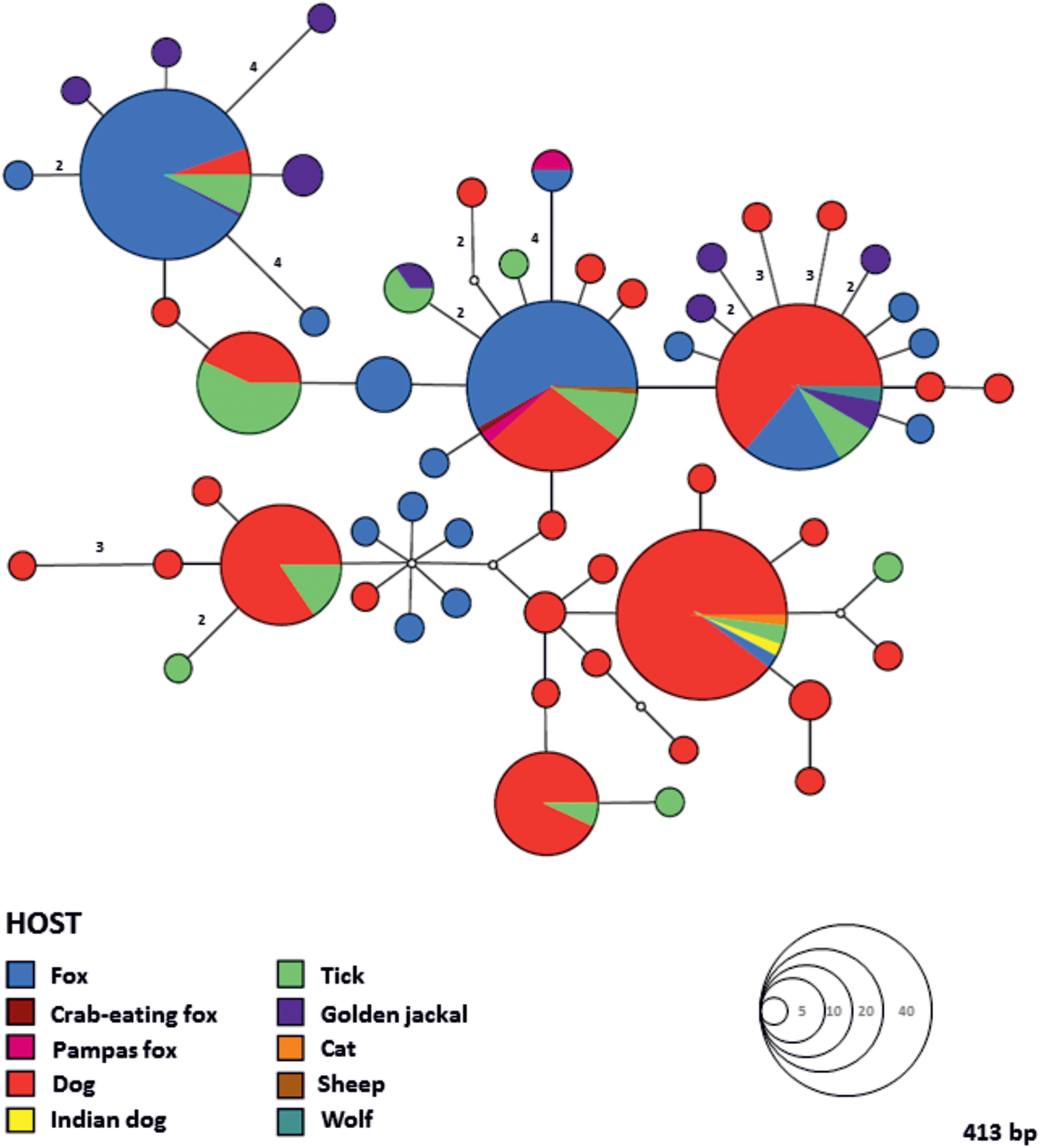
Fig. 2. Median Joining (MJ) haplotype network of the 18S rRNA gene (413 bp) of Hepatozoon canis. Colours indicate different hosts and the circle size in the network corresponds to the number of individuals sharing the same haplotype. Numbers along the branches indicate the number of substitutions between haplotypes.

Fig. 3. Median Joining (MJ) haplotype network of the 18S rRNA gene (413 bp) of Hepatozoon canis. Colours indicate different geographic locality and the circle size in the network denotes the number of individuals sharing the same haplotype. Numbers along the branches indicate the number of substitutions between haplotypes. An asterisk (*) in the circle represents sequences (haplotypes) obtained in the present study.
The representative sequences obtained in the present study have been deposited in GenBank® database and they are available under the following accession numbers: KY693669 (B. canis, 18S rRNA gene), KY693667–KY693668 (Babesia cf. microti, 18S rRNA gene), KY693670 (H. canis, 18S rRNA gene), KY693672 (A. phagocytophilum, 16S rRNA gene), KY693671 (A. phagocytophilum, groEL gene) and KY693673 (B. rochalimae, 16S-23S rRNA gene).
Detection of Hepatozoon canis in fox foetuses
Out of six foetuses retrieved from a H. canis infected vixen, tissues of two (33·3%) tested positive by Hepatozoon-specific PCR. DNA sequencing showed that both foetuses and the respective mother fox were infected with the same haplotype of H. canis, indicating possible intrauterine infection.
DISCUSSION
The present study reports a relatively high overall prevalence and diversity of pathogens transmitted by arthropod vectors in red foxes from western Austria. It also provides an overview of genetic variability, population structures, geographic distribution and host range for the two most frequently detected haematozoa (i.e. Babesia cf. microti and H. canis) in foxes worldwide. Moreover, the results of this study demonstrate, for the first time, the possible transplacental transmission of H. canis from a mother fox to its offspring.
Haematozoan parasites
Three species of apicomplexan haemoparasites namely B. canis, Babesia cf. microti and H. canis were identified in the current study. Babesia cf. microti (order Piroplasmida) is a tick-transmitted parasite affecting dogs and wild carnivores worldwide (Alvarado-Rybak et al. Reference Alvarado-Rybak, Solano-Gallego and Millán2016). It is also known as Theileria annae, Babesia annae, Babesia sp. ‘Spanish dog isolate’ and Babesia vulpes. However, all these names are considered unavailable as the formal description of the agent is still lacking (Harris, Reference Harris2016). The piroplasm is genetically most closely related to zoonotic Babesia microti (Piroplasmida, Babesiidae), therefore in this study we refer to it as ‘Babesia cf. microti’ (cf. is an abbreviation for the Latin word confer which means compare) as recommended by Harris (Reference Harris2016). Babesia cf. microti has been reported to cause severe illness in dogs (Zahler et al. Reference Zahler, Rinder, Schein and Gothe2000; Camacho, Reference Camacho2006; Falkenö et al. Reference Falkenö, Tasker, Osterman-Lind and Tvedten2013; Miró et al. Reference Miró, Checa, Paparini, Ortega, González-Fraga, Gofton, Bartolomé, Montoya, Gálvez, Mayo and Irwin2015), but foxes are considered as major natural reservoir hosts for this species (Otranto et al. Reference Otranto, Cantacessi, Pfeffer, Dantas-Torres, Brianti, Deplazes, Genchi, Guberti and Capelli2015; Alvarado-Rybak et al. Reference Alvarado-Rybak, Solano-Gallego and Millán2016). The pathogen has been found infecting foxes in Europe, Asia and North America (Alvarado-Rybak et al. Reference Alvarado-Rybak, Solano-Gallego and Millán2016) and the prevalence rates range from 0·98% in Italy (Zanet et al. Reference Zanet, Trisciuoglio, Bottero, de Mera, Gortazar, Carpignano and Forroglio2014) to 69·2% in Portugal (Cardoso et al. Reference Cardoso, Cortes, Reis, Rodrigues, Simões, Lopes, Vila-Viçosac, Talmi-Frankd, Eyald, Solano-Gallegoe and Baneth2013). The prevalence of 50·7% (in blood) herein observed is the same as that previously reported in the north-eastern part of Austria (50%; Duscher et al. Reference Duscher, Fuehrer and Kübber-Heiss2014). However, only 36 foxes were examined, representing the only currently existing data on haematozoan parasites in Austrian foxes. The hedgehog tick Ixodes hexagonus (Acari, Ixodidae) was assumed to be the main vector candidate for Babesia cf. microti, since it was the most frequently reported tick species in infected dogs from hyperendemic regions in Spain (Camacho et al. Reference Camacho, Pallas, Gestal, Guitián, Olmeda, Telford and Spielman2003). Nevertheless, the piroplasmid DNA has recently been found in questing Dermacentor reticulatus in Austria, suggesting the possible implication of this tick in the life cycle and transmission of Babesia cf. microti (Hodžić et al. Reference Hodžić, Zörer and Duscher2017). Although the vector competence has not yet been proven for this tick, the existence of the same nucleotide haplotype circulating between foxes and ticks, not only in Austria but all over the Europe, supports this hypothesis.
Another canine babesial species, Babesia canis (Piroplasmida, Babesiidae), was molecularly confirmed in the blood of a single fox. Given that only two cases have been previously reported in foxes in Portugal (Cardoso et al. Reference Cardoso, Cortes, Reis, Rodrigues, Simões, Lopes, Vila-Viçosac, Talmi-Frankd, Eyald, Solano-Gallegoe and Baneth2013) and Bosnia and Herzegovina (Hodžić et al. Reference Hodžić, Alić, Fuehrer, Harl, Wille-Piazzai and Duscher2015), it can be concluded that they are not suitable hosts for B. canis, with hardly any impact as reservoir or spreader (Hodžić et al. Reference Hodžić, Alić, Fuehrer, Harl, Wille-Piazzai and Duscher2015).
Hepatozoon canis (Eucoccidiorida, Hepatozoidae) is a protozoan parasite of domestic and wild carnivores. In general, the geographical distribution of H. canis is related to the distribution of its major tick vector Rhipicephalus sanguineus sensu lato (Acari, Ixodidae), also known as the brown dog tick (Baneth, Reference Baneth2011; Giannelli et al. Reference Giannelli, Lia, Annoscia, Buonavoglia, Lorusso, Dantas-Torres, Baneth and Otranto2017). However, this protozoan has also been reported in areas lacking R. sanguineus and these include Austria (Duscher et al. Reference Duscher, Kübber-Heiss, Richter and Suchentrunk2013, Reference Duscher, Fuehrer and Kübber-Heiss2014), the Czech Republic (Mitková et al. Reference Mitková, Hrazdilová, Steinbauer, D'Amico, Mihalca and Modrý2016), Hungary (Tolnai et al. Reference Tolnai, Sréter-Lancz and Sréter2015) and Slovakia (Majláthová et al. Reference Majláthová, Hurníková, Majláth and Petko2007; Miterpáková et al. Reference Miterpáková, Komjáti-Nagyová, Hurníková and Víchová2017). In addition to the main vector in Europe, a recent study has demonstrated the sporogonic development of H. canis in R. turanicus collected from a naturally infected fox in Italy, confirming its role as a definitive host (Giannelli et al. Reference Giannelli, Lia, Annoscia, Buonavoglia, Lorusso, Dantas-Torres, Baneth and Otranto2017). Hepatozoon canis is one of the most prevalent parasite species found in foxes and the prevalence in Europe reaches up to 77% (Cardoso et al. Reference Cardoso, Cortes, Eyal, Reis, Lopes, Vila-Viçosa, Rodrigues and Baneth2014). The infection rate of H. canis assessed in this study (18·5% blood and 29·8% spleen samples tested positive) is noticeably lower than that recently reported in foxes from north-eastern Austria (58·3%, Duscher et al. Reference Duscher, Fuehrer and Kübber-Heiss2014). Moreover, an extremely low level of genetic variability in the 18S rRNA gene of H. canis, represented by only one haplotype circulating in foxes, was observed in western Austria. Conversely, four haplotypes have been described in 21 foxes in the north-eastern part of the country (Duscher et al. Reference Duscher, Fuehrer and Kübber-Heiss2014), whilst a single golden jackal (Canis aureus) from an area close to Vienna was found bearing six different genetic variants of H. canis (Duscher et al. Reference Duscher, Kübber-Heiss, Richter and Suchentrunk2013). The high mountain ranges (Central Eastern Alps) which separate western Austria i.e. the western provinces of Tyrol and Vorarlberg from the rest of the country could act as a physical geographic barrier inhibiting gene flow and leading to increased inbreeding frequency. All these factors may subsequently contribute to the low degree of genetic variations of H. canis in the studied area. Furthermore, the relatively low prevalence observed in this study compared with other parts of Europe and the existence of only one H. canis haplotype may indicate that the pathogen has recently been introduced into the studied area.
To evaluate which is the most suitable tissue for molecular detection of haematozoa in foxes, blood and spleen were tested and the proportion of positivity was calculated. Our results indicate the blood is better for Babesia cf. microti detection, whereas the spleen is a more sensitive tissue substrate for detection of H. canis DNA, most likely due to the occurrence of merogony and subsequently higher parasite load in the spleen (Cardoso et al. Reference Cardoso, Cortes, Eyal, Reis, Lopes, Vila-Viçosa, Rodrigues and Baneth2014).
Possible evidence for vertical transmission of Hepatozoon canis
Hepatozoon canis has a rather unique transmission mode as it, unlike any other vector-borne pathogen, needs to be ingested by a vertebrate intermediate host. Ingestion of the tick vector (definitive host) containing mature oocysts by the vertebrate host represents a main route of infection (Baneth, Reference Baneth2011). Nevertheless, vertical transmission from infected dogs to their puppies has been demonstrated (Murata et al. Reference Murata, Inoue, Tateyama, Taura and Nakama1993). This non-vectorial way of infection has been hypothesized to occur in foxes, but it has never been confirmed. In this study, we detected H. canis DNA in tissues of two out of six foetuses retrieved from an infected vixen, and the same nucleotide sequence was confirmed in both foetuses and the mother fox, indicating the possible intrauterine infection. The existence of a transplacental route of infection in foxes could explain the high prevalence of H. canis in an area, including Austria, where Rhipicephalus tick vectors are not present. It would thus seem that female foxes play an important role in the maintenance of H. canis, as has already been suggested (Hodžić et al. Reference Hodžić, Alić, Fuehrer, Harl, Wille-Piazzai and Duscher2015).
Arthropod-borne bacteria and zoonotic implications
In recent years, bacteria transmitted by blood-feeding arthropods have attracted the attention of many researchers since they were recognized as important causative agents of human and animal diseases (Hodžić et al. Reference Hodžić, Alić, Fuehrer, Harl, Wille-Piazzai and Duscher2015). Among wild carnivores, foxes display a limited impact on the circulation of emerging zoonotic bacteria such as A. phagocytophilum, Borrelia burgdorferi sensu lato and B. rochalimae due to very low infection rates, but they may still represent a potential source of human infections (Henn et al. Reference Henn, Chomel, Boulouis, Kasten, Murray, Bar-Gal, King, Courreau and Baneth2009; Dumitrache et al. Reference Dumitrache, Matei, Ionică, Kalmár, D'Amico, Sikó-Barabási, Ionescu, Gherman and Mihalca2015). Results from our study could support this as four foxes were infected with the same strains of A. phagocytophilum and B. rochalimae previously reported in human patients with apparent signs of illness (Petrovec et al. Reference Petrovec, Sumner, Nicholson, Childs, Strle, Barlic, Lotric-Furlan and Avsic Zupanc1999; Eremeeva et al. Reference Eremeeva, Gerns, Lydy, Goo, Ryan, Mathew, Ferraro, Holden, Nicholson, Dasch and Koehler2007; Markowicz et al. Reference Markowicz, Schötta, Wijnveld and Stanek2016). However, data on the prevalence of A. phagocytophilum and B. rochalimae in foxes in Europe is scant and limited to very few countries such as the Czech Republic (Hulínská et al. Reference Hulínská, Langrová, Pejcoch and Pavlásek2004), Germany (Härtwig et al. Reference Härtwig, von Loewenich, Schultze, Straubinger, Daugschies and Dyachenko2014), Italy (Ebani et al. Reference Ebani, Verin, Fratini, Poli and Cerri2011), Poland (Karbowiak e t al. Reference Karbowiak, Víchová, Majláthová, Hapunik and Peťko2009), Romania (Dumitrache et al. Reference Dumitrache, Matei, Ionică, Kalmár, D'Amico, Sikó-Barabási, Ionescu, Gherman and Mihalca2015) and France (Henn et al. Reference Henn, Chomel, Boulouis, Kasten, Murray, Bar-Gal, King, Courreau and Baneth2009).
Moreover, two foxes were found to be positive for a potentially new species of a recently specified cluster Candidatus Neoehrlichia, which is phylogenetically more closely related to a raccoon-associated species Candidatus Neoehrlichia lotoris from North America, than to zoonotic Candidatus Neoehrlichia mikurensis (data published in Hodžić et al. Reference Hodžić, Cézanne, Duscher, Harl, Glawischnig and Fuehrer2015a ). The same sequences of Candidatus Neoehrlichia sp. (16S rRNA and groEL genes) were found later in a fox from the Czech Republic (Hodžić et al. Reference Hodžić, Mitkovà, Modrý, Juránková, Frgelecová, Forejtek, Steinbauer and Duscher2017a ) and in an European badger (Meles meles) from Hungary (Hornok et al. Reference Hornok, Trauttwein, Takács, Hodžić, Duscher and Kontschán2017), expanding our knowledge on geographic distribution and host association for this enigmatic bacterium. Regarding rickettsial species from the spotted fever group, they have been molecularly confirmed in arthropods collected from foxes (Marié et al. Reference Marié, Davoust, Socolovschia, Mediannikova, Roqueplo, Beaucournu, Raoult and Parola2012; Torina et al. Reference Torina, Blanda, Antoci, Scimeca, D'Agostino, Scariano, Piazza, Galluzzo, Giudice and Caracappa2013), but never in this animal species itself (Duscher et al. Reference Duscher, Fuehrer and Kübber-Heiss2014; Hodžić et al. Reference Hodžić, Alić, Fuehrer, Harl, Wille-Piazzai and Duscher2015), which opens the question about the ability of foxes to serve as hosts for these emerging zoonotic bacteria. However, most of the studies conducted on foxes used either blood or spleen for molecular detection of rickettsial DNA, but other tissues (e.g. skin) might be more suitable for detection consistency, as seen in guinea pigs experimentally infected with Rickettsia rickettsii (Zemtsova et al. Reference Zemtsova, Montgomery and Levin2015). Large-scale studies employing several gene loci, more sensitive diagnostic tools and/or even in vitro culturing are needed to shed more light on the possible involvement of foxes in the transmission cycles of zoonotic bacteria transmitted by arthropods.
Filarial nematodes and Leishmania infantum
Neither blood nor spleen samples from animals tested in this study were PCR positive for filarial nematodes and Leishmania species. In general, Dirofilaria immitis and D. repens as well as Leishmania infantum have been molecularly confirmed in foxes in Europe (Magi et al. Reference Magi, Calderini, Gabrielli, Dell'Omodarme, Macchioni, Prati and Cancrini2008; Penezić et al. Reference Penezić, Selaković, Pavlović and Ćirović2014; Tolnai et al. Reference Tolnai, Széll, Sproch, Szeredi and Sréter2014; Cardoso et al. Reference Cardoso, Gilad, Cortes, Nachum-Biala, Lopes, Vila-Viçosa, Simões, Rodrigues and Baneth2015; Karayiannis et al. Reference Karayiannis, Ntais, Messaritakis, Tsirigotakis, Dokianakis and Antoniou2015), however their impact in the transmission of these zoonotic parasites still remains unclear. Infection with D. repens has occasionally been reported in dogs and humans (Fuehrer et al. Reference Fuehrer, Auer, Leschnik, Silbermayr, Duscher and Joachim2016), and mosquitoes in Eastern Austria (Silbermayr et al. Reference Silbermayr, Eigner, Joachim, Duscher, Seidel, Allerberger, Indra, Hufnagl and Fuehrer2014). Nevertheless, L. infantum DNA was recently detected in Phlebotomus mascittii sand flies and in a dog from Austria (Obwaller et al. Reference Obwaller, Karakus, Poeppl, Töz, Özbel, Aspöck and Walochnik2016). All these findings may indicate that the infections, in particular those caused by D. repens became endemic in this country (Fuehrer et al. Reference Fuehrer, Auer, Leschnik, Silbermayr, Duscher and Joachim2016). However, further monitoring of animals and arthropod-vectors from all regions in Austria is required in order to investigate the distribution and potential risk of infection.
Concluding remarks
This work represents the first comprehensive molecular study on pathogens transmitted by blood-feeding arthropods in Austrian foxes, and the results obtained reveal the frequency of occurrence and diversity of protozoan and bacterial agents in the sampled area. Moreover, this study further demonstrates that foxes may play a pivotal role in maintaining, circulating and spreading of Babesia cf. microti and H. canis. However, none of these haematozoan parasites have yet been diagnosed in dogs in Austria, most likely because they could easily have been overlooked, due to a very low parasitaemia and absence of specific clinical signs (Mitková et al. Reference Mitková, Hrazdilová, Steinbauer, D'Amico, Mihalca and Modrý2016). Molecular diagnostic tools are therefore to be highly recommended for the detection and identification of these parasites in domestic dogs and other wild carnivores as potential hosts for these agents.
ACKNOWLEDGMENT
The authors would like to thank Barbara Eigner (University of Veterinary Medicine Vienna, Austria) for her help in laboratory work, and to Prof. Dr David Modrý and Barbora Mitková (University of Veterinary and Pharmaceutical Sciences, Brno, Czech Republic) for helpful discussions and support. The work of Adnan Hodžić, Georg G. Duscher and Hans-Peter Fuehrer was conducted under the frame of EurNegVec COST Action TD1303.
FINANCIAL SUPPORT
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.





