Introduction
Nutrition is directly responsible for the delivery of energy for cellular metabolism, as well as providing the diverse array of substrates required for normal physiological function. It also plays a central role in the modulation of inflammatory and disease processes(Reference Bosma-den Boer, van Wetten and Pruimboom1–Reference Kopp3) and, thus, can be utilised as a therapeutic intervention. Nutritional therapies that limit substrate availability and produce ketosis (such as fasting, calorie restriction and ketogenic diets) directly impact metabolism and cellular energetics(Reference Shen, Kapfhamer and Minnella2,Reference Ruskin and Masino4) . Ketosis has been reported to be effective in neurological conditions characterised by neurodegeneration(Reference Davis, Fournakis and Ellison5–Reference Taylor, Sullivan, Mahnken, Burns and Swerdlow9), psychological disorders(Reference Morris, Puri and Carvalho10), brain injury(Reference Camberos-Luna and Massieu6,Reference McDougall, Bayley and Munce11) and nervous system excitability(Reference Li, Liu, Liu and Li12–Reference Mahmoud, Ho-Huang and Buhler15). More recently, the presence of ketones has been suggested to influence pain mechanisms(Reference Ruskin, Kawamura and Masino16–Reference Masino and Ruskin18). Given this, ketosis produced through a ketogenic diet may be an appropriate treatment strategy for persistent pain, a dysfunction within the nervous system involving changes in both cortical structure and function(Reference Schabrun, Elgueta-Cancino and Hodges19,Reference Schabrun, Christensen, Mrachacz-Kersting and Graven-Nielsen20) . Neuroplastic remodelling facilitates increased connectivity and amplification of pain perception and is required to shift into a persistent pain state(Reference Kuner and Flor21). Broadly, nutritional interventions have been shown to improve pain outcomes(Reference Elma, Yilmaz and Deliens22–Reference Field, Pourkazemi, Turton and Rooney24). Directly targeting neurobiology through a ketogenic diet could potentially modulate maladaptive change and become an additional strategy to add to comprehensive chronic pain management(Reference Nijs, Elma and Yilmaz25).
The concept of nutritional neurobiology for chronic pain management is starting to appear in the literature, where dietary intake can be both a trigger for upregulated pain mechanisms but also potentially provide therapeutic options(Reference Nijs, Elma and Yilmaz25,Reference Kaushik, Strath and Sorge26) . There have been three systematic reviews published to date(Reference Elma, Yilmaz and Deliens22–Reference Field, Pourkazemi, Turton and Rooney24) that report outcomes on human participants with chronic pain from dietary interventions, all published in the last 2 years. These reviews report the effectiveness of improved nutrition generally as a pain management option, particularly when considering nutrient-dense whole-food diets and the removal of discretionary ultra-processed foods high in sugar and fat. They are unable to clearly point to any specific diet as the best treatment, however. A recent review suggested both the Mediterranean diet and a carbohydrate-restricted diet were promising diets for reducing the impact of chronic pain by either a reduction in inflammation or a reduction in oxidative stress(Reference Kaushik, Strath and Sorge26). The authors note, however, that only two studies reviewed were specifically assessing the context of chronic pain (knee osteoarthritis) and the rest were examining participants with metabolic dysregulation (such as elevated cardiovascular risk or obesity).
Nutritional ketosis is achieved through a ketogenic diet by restricting dietary carbohydrates sufficiently to shift cellular energetics from glucose to fat oxidation as the main fuel source(Reference Hite, Cavan and Cywes27). Ketone bodies (β-hydroxybutyrate and acetoacetate) are produced in the liver (ketosis) and delivered via the bloodstream as part of this alternate fuel pathway, providing both a fuel source and a signalling molecule that can modulate many physiological processes(Reference Camberos-Luna and Massieu6). As a signalling molecule, β-hydroxybutyrate is a metabolic intermediary that can act as an endogenous class I and II histone deacetylase inhibitor involved in the regulation of longevity and antioxidant defences, diseases of aging, and also diabetes and cancer(Reference Morris, Puri and Carvalho10,Reference Newman and Verdin28,Reference Shimazu, Hirschey and Newman29) . It acts as a ligand for G-protein-coupled receptors (hydroxycarboxylic acid receptor 2) and free fatty acid receptor 3, which bind short-chain fatty acids, regulate metabolism and play a role in the development of metabolic disease states(Reference Newman and Verdin28). Ketone signalling via a ketogenic diet has been reported to beneficially effect physiological processes involved in many disease conditions, including obesity, cancer, diabetes, epilepsy, Parkinson’s disease, Alzheimer’s disease, multiple sclerosis, peripheral neuropathy, liver disease, inherited metabolic disorders, muscle degeneration, polycystic ovarian syndrome, irritable bowel syndrome, migraine and fibromyalgia(Reference Li, Liu, Liu and Li12,Reference Eendfeldt and Scher30) .
Whilst ketogenic diets have reported efficacy clinically for humans in a variety of neurological conditions, the evidence for plausible physiological mechanisms by which the nervous system may be modulated relies heavily on animal models (both in vivo and in vitro) to explore the mechanistic pathways. The mechanisms suggested are neuroprotective and neuromodulatory, whereby decreasing glycolytic metabolism and shifting to fat oxidation raises ATP and adenosine levels and improves cellular energetics. It also activates multiple signalling pathways involved in the reduction of reactive oxygen species in neurological tissues, increased mitochondrial number and function, synaptic regulation, and inhibition of pro-inflammatory cytokine mediators(Reference Masino and Ruskin18,Reference Paoli, Rubini, Volek and Grimaldi31–Reference Ruskin and Masino35) . The overall effect would seem to be restoring homeostatic synaptic function and excitability.
To date, there is limited published literature on human clinical trials that examine a ketogenic diet as a treatment for chronic pain. The purpose of this scoping review was to investigate animal models that report outcomes related to the nervous system by changing from a standard animal diet to a ketogenic diet. It includes multiple models of nervous system dysfunction and synthesises the outcomes presented into broader themes by which a ketogenic diet may plausibly modulate biological pathways associated with human chronic pain perception. It also discusses the potential issues with clinical translation from animal models to human models of dietary interventions.
Methods
Protocol
The framework for this review was based on relevant items of the scoping review protocol and PRISMA-ScR checklist from the Joanna Briggs Institute(Reference Peters, Godfrey, McInerney, Aromataris and Munn36,Reference Tricco, Lillie and Zarin37) to answer the research question: ‘How does a ketogenic diet in animal models influence the nervous system?’
Eligibility criteria
Studies were included if they met the following criteria:
-
1. Mammal models that report an ad libitum high-fat, low-carbohydrate ketogenic diet that is ≥7 d (% energy from fat ≥69 % or 3:1 ratio of fat:protein + carbohydrate + fibre + extras) as the intervention. The minimum diet length of 1 week was used to ensure all studies were captured, and was based on similar systematic reviews reporting studies where the minimum reported length of diet was 2 weeks in rat and mouse models assessing both metabolic and nervous system outcomes(Reference Campbell, Senior and Bell-Anderson38,Reference Auvinen, Romijn and Biermasz39) .
-
2. Studies that report objective outcomes related to nervous system function including neuroinflammation.
-
3. Experimental study designs: longitudinal pre–post intervention trials including randomised controlled trials.
Studies were excluded if:
-
1. The diets were both high in fat and carbohydrate, carbohydrate levels exceeded 10 % or where the chow was not described, and the ketogenic status could not be confirmed.
-
2. The diet was not ad libitum or provided in the form of whole food, including oral gavage, intraperitoneal models, food extracts or exogenous ketones.
-
3. The subjects were human or in vitro cultures.
-
4. The model used represented cancer or genetic syndromes.
-
5. The paper was not in English.
Information sources and search strategy
An electronic database search including Medline, EMBASE, Cochrane Library for controlled trials, AMED via OVID, CINAHL via Ebsco, Web of Science and PubMed was carried out on 5 July 2020 and included dates from database inception to the search date. A preliminary search refined the search strategy, with the key terms outlined in Supplementary Table 1. Additional searches included a Google Scholar search to check identified articles ‘cited by’ and ‘related articles’ links, and reference checks on identified articles with subsequent hand search for these and inclusion if they met the criteria. Retrieved references were downloaded into EndNote reference management software (Endnote X7.7.1, Thomson Reuters 2016) and then imported using Covidence systematic review software (Veritas Health Innovation, Melbourne, Australia).
Study selection and screening
Duplicates were removed, then titles and abstracts were assessed in Covidence by two reviewers independently (R.F. and T.F) against the eligibility criteria. Full texts of identified studies were then screened by two reviewers independently (R.F. and T.F) for final eligibility, with any disagreements resolved by a third reviewer.
Data items
The primary outcomes of interest were changes in nervous system function (such as excitability) or energetics (such as altered substrate measured by blood glucose or ketones levels) that report a plausible biological mechanism by which a ketogenic diet may influence the nervous system. Additional data extracted included: author, year of study, animal, animal variant, chow ratios, disease model and intervention diet length. Critical appraisal of the literature to assess risk of bias was not carried out due to the frequently poor-quality methods employed in animal research. This includes lack of randomisation, lack of blinding and incorrect statistical methods(Reference Hooijmans and Ritskes-Hoitinga40,Reference Pound and Ritskes-Hoitinga41) . Nervous system outcomes were taken as presented by the study authors.
Data charting process and synthesis of results
Data items were extracted and compiled in an excel spreadsheet. Primary outcomes were reviewed, and a subjective thematic analysis was carried out by R.F. The process of thematic analysis involved building a list of categories that best fit the outcome description given by the study author. These were then further synthesised into broad themes. Studies with more than one relevant theme could be allocated into more than one theme. A random sample of thirty-five studies (20 %) was independently reviewed by T.F to ensure consistency of theme allocation.
Results
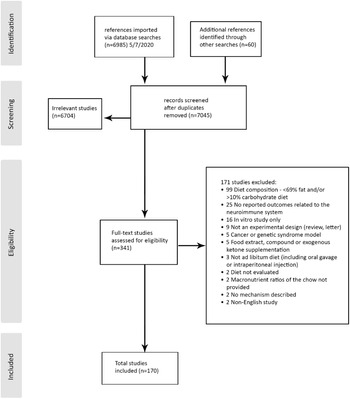
A systematic search of the databases retrieved 7045 studies screened for eligibility after duplicates were removed. A total of 341 full-text articles were assessed with a total of 170 meeting the inclusion criteria and included in the scoping review (Fig. 1). Of the ninety-nine studies excluded for being either >10 % carbohydrate or ≤69 % fat, only three studies were described as lower in ketogenic (but still included 32 % carbohydrate, 20 % carbohydrate or 30 % fat). The remaining studies were captured by the search term ‘high fat’, which retrieved studies high in both fat and carbohydrate designed to produce obesity or metabolic dysfunction.

Fig. 1. Inclusion flowchart.
Characteristics of included studies
The studies comprised 103 rat studies, 63 mouse studies, 2 that included rats and mice, and 1 that included rats and gerbils as well as a canine case study. There was a range of nervous system dysfunction models, including five age-related degeneration(Reference Hernandez, Hernandez and Campos42–Reference Zhang, Xu, Kerwin, LaManna and Puchowicz46), four Alzheimer’s disease(Reference Beckett, Studzinski, Keller, Paul Murphy and Niedowicz47–Reference Van der Auwera, Wera, Van Leuven and Henderson50), seven autism(Reference Ahn, Narous, Tobias, Rho and Mychasiuk51–Reference Smith, Rho and Teskey57), four cerebral ischaemia(Reference Tai and Truong58–Reference Yang, Guo and Wang61), two pain perception(Reference Ruskin, Kawamura and Masino16,Reference Ruskin, Suter, Ross and Masino62) , twenty-four general central nervous system(Reference Elamin, Ruskin, Masino and Sacchetti63–Reference Ziegler, Ribeiro and Hagenn86), two diabetes(Reference Morrison, Hill and DuVall87,Reference Yamada, Rensing and Thio88) , ninety-one epilepsy(Reference Bough and Eagles89–Reference Masino, Freedgood and Reichert179), two metabolic syndrome(Reference Kephart, Mumford and Mao180,Reference Mohamed, El-Swefy, Rashed and Abd El-Latif181) , one mild cognitive impairment(Reference Hargrave, Davidson, Lee and Kinzig182), two multiple sclerosis(Reference Kim, Hao and Liu183,Reference Stumpf, Berghoff and Trevisiol184) , one nerve toxin(Reference Myers and Langston185), four optic nerve dysfunction(Reference Bernardo-Colon, Vest and Clark186–Reference Zarnowski, Choragiewicz and Schuettauf189), two Parkinson’s disease(Reference Cheng, Yang and An190,Reference Yang and Cheng191) , three peripheral nerve dysfunction(Reference Cooper, McCoin and Pei192–Reference Liskiewicz, Wlaszczuk and Gendosz194), four spinal cord injury(Reference Kong, Huang and Ji195–Reference Wang, Wu and Liu198), three stroke(Reference Guo, Wang and Zhao199–Reference Rahman, Muhammad and Khan201) and nine traumatic brain injury(Reference Deng-Bryant, Prins, Hovda and Harris202–Reference Zhang, Wu, Jin and Zhang210). The length of the dietary intervention ranged from 1 week to 6 months.
Fourteen broad themes involving nervous system function were identified. These themes, the disease models used, and further details are presented in Table 1. Detailed information on individual study characteristics and reported outcomes is compiled in Supplementary Table 2, which references all 170 included studies.
-
1. Alterations in cellular energetics and metabolism (reported in twenty-eight studies across nine disease models (Reference Hernandez, Hernandez and Campos42,Reference Hernandez, Hernandez and Campos43,Reference Zhang, Xu, Kerwin, LaManna and Puchowicz46,Reference Roy, Nugent and Tremblay-Mercier49,Reference Elamin, Ruskin, Masino and Sacchetti63,Reference Leino, Gerhart, Duelli, Enerson and Drewes68,Reference Melo, Nehlig and Sonnewald70,Reference Pifferi, Tremblay and Croteau72,Reference Roy, Beauvieux and Naulin74–Reference Selfridge, Wilkins and Lezi76,Reference Wang, Liu, Zhou, Wu and Zhu83,Reference Zhang, Zhang, Marin-Valencia and Puchowicz85,Reference Morrison, Hill and DuVall87,Reference Bough, Matthews and Eagles91,Reference Bough, Wetherington and Hassel95,Reference Bough97,Reference Forero-Quintero, Deitmer and Becker108,Reference Kawamura, Ruskin, Geiger, Boison and Masino125,Reference Mantis, Meidenbauer, Zimick, Centeno and Seyfried136,Reference Nakazawa, Kodama and Matsuo142,Reference Samala, Willis and Borges156,Reference Harun-Or-Rashid, Pappenhagen and Palmer187,Reference Harun-Or-Rashid and Inman188,Reference Cooper, McCoin and Pei192,Reference Streijger, Plunet and Lee197,Reference Deng-Bryant, Prins, Hovda and Harris202,Reference Prins and Hovda207) ). The reduced glucose consumption of a ketogenic diet resulted in lower glucose availability within the nervous system and a shift to fat-based metabolism with up-regulation of processes required to deliver this alternate energy substrate. Fat-based metabolism was reported to improve energy availability, utilisation and efficiency. It was also reported to reduce low-grade inflammation driven by a low energy state.
-
2. Biochemical (reported in three studies in epilepsy models (Reference Chwiej, Patulska and Skoczen102–Reference Chwiej, Patulska and Skoczen104) ). Elemental changes (P, S, K, Ca, Fe, Cu, Zn and Se) within the hippocampus were assessed via X-ray fluorescence microscopy with significant changes, with a significant decrease in P, K and Zn, and a significant increase in Ca and Se as a result of the ketogenic diet. As hippocampal levels of Ca increase with seizures, these changes did not provide evidence supporting a mechanism for seizure reduction. Additionally, the ratio of absorbance for specific biological macromolecules (such as ketones and lipids) was increased with the possibility of these molecules being involved in anti-seizure mechanism rather than elemental changes.
-
3. Cortical/neuronal excitability (reported in fifty-three studies of which forty-nine were epilepsy models (Reference Dai, Zhao and Tomi53,Reference Smith, Rho and Teskey57,Reference Huang, Li and Wu67,Reference Bough and Eagles89–Reference Bough, Schwartzkroin and Rho94,Reference Chwiej, Patulska and Skoczen104,Reference Dutton, Sawyer and Kalume107,Reference Gama, Trindade-Filho and Oliveira109–Reference Godlevskii, Polyasny and Ovchinnikova111,Reference Hansen, Nielsen and Knudsen113–Reference Hartman, Zheng, Bergbower, Kennedy and Hardwick116,Reference Hori, Tandon, Holmes and Stafstrom118,Reference Jiang, Yang and Wang124,Reference Kawamura, Ruskin, Geiger, Boison and Masino125,Reference Kobow, Kaspi and Harikrishnan127,Reference Kresyun, Polyasny, Godovan and Godlevsky129,Reference Likhodii, Musa and Mendonca131,Reference Lusardi, Akula and Coffman135,Reference Melo, Rego and Bueno140–Reference Nakazawa, Kodama and Matsuo142,Reference Noh, Kim and Lee144,Reference Nylen, Likhodii, Abdelmalik, Clarke and Burnham150–Reference de Almeida Rabello Oliveira, da Rocha Ataíde and de Oliveira152,Reference Raffo, Francois, Ferrandon, Koning and Nehlig154–Reference Samala, Willis and Borges156,Reference Simeone, Wilke and Milligan158–Reference Szot, Weinshenker, Rho, Storey and Schwartzkroin164,Reference Thavendiranathan, Mendonca and Dell166,Reference Thavendiranathan, Chow, Cunnane and Burnham167,Reference Todorova, Tandon, Madore, Stafstrom and Seyfried170–Reference Wang, Hou and Lu173,Reference Zarnowska, Luszczki and Zarnowski175,Reference Ziegler, Oliveira and Pires177–Reference Masino, Freedgood and Reichert179,Reference Schwartzkroin, Wenzel and Lyeth209) ). The ketogenic diet was broadly reported to restore the balance of nervous system excitability toward homeostatic levels; however, some studies reported neutral or negative findings(Reference Bough, Matthews and Eagles91,Reference Bough, Gudi, Han, Rathod and Eagles93,Reference Melo, Rego and Bueno140,Reference Nylen, Likhodii, Abdelmalik, Clarke and Burnham150,Reference Nylen, Likhodii, Hum and Burnham151,Reference Raffo, Francois, Ferrandon, Koning and Nehlig154,Reference Thavendiranathan, Mendonca and Dell166,Reference Zarnowska, Luszczki and Zarnowski175,Reference Blaise, Ruskin, Koranda and Masino178) . This category was largely composed of epilepsy models that described reductions in frequency, threshold, duration, latency and spread of seizures. Restoration of circadian rhythms within the brain was also reported.
-
4. Epigenetic regulation (reported in thirty studies across nine disease models (Reference Hernandez, Hernandez and Truckenbrod44,Reference Newell, Shutt and Ahn55,Reference Yang, Guo and Wang61,Reference Ling, Wang, Sun, Zhao and Ni69,Reference Pifferi, Tremblay and Croteau72,Reference Selfridge, Wilkins and Lezi76,Reference Bough, Wetherington and Hassel95,Reference Bough97,Reference Cheng, Kelley and Wang99,Reference Cheng, Hicks, Wang, Eagles and Bondy100,Reference Cullingford, Eagles and Sato105,Reference Dupuis, Curatolo, Benoist and Auvin106,Reference Forero-Quintero, Deitmer and Becker108,Reference Jeong, Kim and Kim122,Reference Kobow, Kaspi and Harikrishnan127,Reference Lin, Lu and Zeng132,Reference Lusardi, Akula and Coffman135, Reference Noh, Lee and Kim145,Reference Noh, Kang and Kim146,Reference Noh, Kim and Kang149,Reference Silva, Rocha and Pires157,Reference Tabb, Szot, White, Liles and Weinshenker165,Reference Wang, Ding and Ding172,Reference Xu, Sun and Jin174,Reference Ziegler, Araujo, Rotta, Perry and Goncalves176,Reference Ziegler, Oliveira and Pires177,Reference Mohamed, El-Swefy, Rashed and Abd El-Latif181,Reference Hargrave, Davidson, Lee and Kinzig182,Reference Cooper, McCoin and Pei192,Reference Salberg, Weerwardhena, Collins, Reimer and Mychasiuk208) ). The genes reportedly altered by the ketogenic diet generally pertained to the disease model being investigated. Overall, they tended to up-regulate beneficial genetic expression regarding neuroinflammation, neurodegeneration and neuroprotection.
-
5. Mitochondrial function (reported in eighteen studies across ten disease models (Reference Lauritzen, Hasan-Olive and Regnell45,Reference Ahn, Narous, Tobias, Rho and Mychasiuk51,Reference Ahn, Sabouny and Villa52,Reference Selfridge, Wilkins and Lezi76,Reference Sullivan, Rippy and Dorenbos78,Reference Bough97,Reference Hasan-Olive, Lauritzen and Ali117,Reference Jarrett, Milder, Liang and Patel120,Reference Luan, Zhao, Zhai, Chen and Li134,Reference Wang, Hou and Lu173,Reference Kephart, Mumford and Mao180,Reference Stumpf, Berghoff and Trevisiol184,Reference Harun-Or-Rashid, Pappenhagen and Palmer187,Reference Cooper, McCoin and Pei192,Reference Guo, Wang and Zhao199,Reference Greco, Glenn, Hovda and Prins203–Reference Hu, Wang and Qiao205) ). The overall reported benefit to the mitochondria within the nervous system was positive, with increases in number, and improvements in structure and function including energy production and redox balance.
-
6. Neuroinflammation (reported in seven studies across six disease models (Reference Jeong, Jeon and Shin123,Reference Kim, Hao and Liu183,Reference Harun-Or-Rashid and Inman188,Reference Yang and Cheng191,Reference Guo, Wang and Zhao199,Reference Hu, Wang, Jin and Yin204,Reference Prins, Fujima and Hovda206) ). Ketones were reported to inhibit the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome expressed in the nervous system and subsequent reduction of the downstream inflammatory signalling pathways it generates. Neuroinflammation was also reportedly reduced through a reduction in reactive oxygen species. Neuroinflammation was frequently reported in terms of signalling pathways, so many of the relevant studies were reported in theme 12.
-
7. Neuroplasticity and structural integrity (reported in twenty-six studies across ten disease models (Reference Lauritzen, Hasan-Olive and Regnell45,Reference Beckett, Studzinski, Keller, Paul Murphy and Niedowicz47,Reference Van der Auwera, Wera, Van Leuven and Henderson50,Reference Mychasiuk and Rho54,Reference Rho, Sarnat, Sullivan, Robbins and Kim73,Reference Strandberg, Kondziella, Thorlin and Asztely77,Reference Sussman, Germann and Henkelman79,Reference Thio, Rensing and Maloney80,Reference Jiang, Yang and Wang124,Reference Kwon, Jeong, Kim, Choi and Son130,Reference Linard, Ferrandon, Koning, Nehlig and Raffo133,Reference Luan, Zhao, Zhai, Chen and Li134,Reference Muller-Schwarze, Tandon and Liu141,Reference Ni, Zhao and Tian143,Reference Noh, Kim and Lee144,Reference Noh, Kim, Kang, Cho and Choi147,Reference Kim, Hao and Liu183,Reference Stumpf, Berghoff and Trevisiol184,Reference Bernardo-Colon, Vest and Clark186,Reference Zarnowski, Choragiewicz and Schuettauf189,Reference Cooper, Menta and Perez-Sanchez193,Reference Liskiewicz, Wlaszczuk and Gendosz194,Reference Streijger, Plunet and Lee197,Reference Guo, Wang and Zhao199–Reference Rahman, Muhammad and Khan201) ). Improved synaptic plasticity (long-term potentiation) and a reduction of maladaptive plasticity (such as mossy fibre sprouting in epilepsy models) was reported on a ketogenic diet. Other structural changes reported across a range of disease models included: improved myelin formation, reduced axonal degeneration, improved white matter development, reduction in β-amyloid, increased neuronal progenitor cells following seizure, prevention of neuronal loss in the ipsilateral hippocampus, reversal of hippocampal atrophy and lesions, improved neuronal recovery following insult when the diet commenced pre-injury, and reduction in retinal ganglion cell loss.
-
8. Neuroprotection (reported in six studies across four disease models (Reference Tai and Truong58,Reference Tai, Nguyen, Pham and Truong59,Reference Yamada, Rensing and Thio88,Reference Myers and Langston185,Reference Cheng, Yang and An190,Reference Yang and Cheng191) ). A reduction of neuronal apoptosis and neuronal death was reported as a result of the diet in a variety of disease models. Protection against seizure was commonly reported in the epilepsy models and reported in theme 3.
-
9. Neurotransmitter function (reported in ten studies across four disease models (Reference Lauritzen, Hasan-Olive and Regnell45,Reference Fukushima, Ogura and Furuta64,Reference Melo, Nehlig and Sonnewald70,Reference Roy, Beauvieux and Naulin74,Reference Zhang, Zhang, Marin-Valencia and Puchowicz85,Reference Bough, Paquet and Pare96,Reference Calderón, Betancourt, Hernández and Rada98,Reference Church, Adams and Wyss101,Reference Olson, Vuong and Yano153,Reference Cheng, Yang and An190) ). Various mechanisms around neurotransmitter production and clearance were reported with proposed benefit from the diet being an improved GABA levels or GABA-to-glutamate ratio. Parkinson’s disease models reported improvements around dopamine levels.
-
10. Nociception (reported in three studies, two for pain and one for peripheral nerve dysfunction (Reference Ruskin, Kawamura and Masino16,Reference Ruskin, Suter, Ross and Masino62,Reference Cooper, Menta and Perez-Sanchez193) ). A reduction in both allodynia and thermal pain sensitivity was reported that was not dependent on lowered glucose levels.
-
11. Redox balance (reported in fifteen studies across eight disease models (Reference Elamin, Ruskin, Masino and Sacchetti63,Reference Milder, Liang and Patel71,Reference Sullivan, Rippy and Dorenbos78,Reference Ziegler, Ribeiro and Hagenn86,Reference Bough, Yao and Eagles92,Reference Jarrett, Milder, Liang and Patel120,Reference Kephart, Mumford and Mao180,Reference Mohamed, El-Swefy, Rashed and Abd El-Latif181,Reference Bernardo-Colon, Vest and Clark186,Reference Cheng, Yang and An190,Reference Cooper, McCoin and Pei192,Reference Kong, Huang and Ji195,Reference Lu, Yang and Zhou196,Reference Greco, Glenn, Hovda and Prins203,Reference Zhang, Wu, Jin and Zhang210) ). Several studies found an improvement in redox balance through either a reduction in nervous system reactive oxygen species or an increase in antioxidant defence.
-
12. Signalling pathways (reported in thirty-six studies across eight disease models (Reference Ma, Wang and Parikh48,Reference Mychasiuk and Rho54,Reference Newell, Johnsen and Yee56,Reference Tai, Pham and Truong60,Reference Yang, Guo and Wang61,Reference Elamin, Ruskin, Masino and Sacchetti63,Reference Genzer, Dadon, Burg, Chapnik and Froy65,Reference Heischmann, Gano and Quinn66,Reference Ling, Wang, Sun, Zhao and Ni69,Reference Milder, Liang and Patel71,Reference Vizuete, de Souza and Guerra82,Reference Zarnowski, Choragiewicz and Tulidowicz-Bielak84,Reference Gomez-Lira, Mendoza-Torreblanca and Granados-Rojas112,Reference Hu, Cheng, Fei and Xiong119,Reference Jeon, Lee and Kim121,Reference Jeong, Jeon and Shin123,Reference Kawamura, Ruskin, Geiger, Boison and Masino125,Reference Knowles, Budney and Deodhar126,Reference Likhodii, Musa and Mendonca131,Reference Lusardi, Akula and Coffman135,Reference Martillotti, Weinshenker, Liles and Eagles137–Reference McDaniel, Rensing, Thio, Yamada and Wong139,Reference Ni, Zhao and Tian143,Reference Noh, Kim, Cho, Choi and Kang148,Reference Noh, Kim and Kang149,Reference Simeone, Matthews, Samson and Simeone161,Reference Szot, Weinshenker, Rho, Storey and Schwartzkroin164,Reference Tian, Ni and Sun168,Reference Tian, Li, Zhang and Ni169,Reference Harun-Or-Rashid, Pappenhagen and Palmer187,Reference Lu, Yang and Zhou196,Reference Wang, Wu and Liu198–Reference Rahman, Muhammad and Khan201) ). A variety of signalling pathways were reported depending on the disease model being used. These centred around other key mechanisms such as reduced neuroinflammation, reduced oxidative stress, altered neuronal energy metabolism, reduced cortical excitability and reduced neurodegeneration.
-
13. Synaptic transmission (reported in seven studies across three disease models (Reference Hernandez, Hernandez and Campos42,Reference Hernandez, Hernandez and Truckenbrod44,Reference Huang, Li and Wu67,Reference Bough, Wetherington and Hassel95,Reference Bough97,Reference Koranda, Ruskin, Masino and Blaise128,Reference Blaise, Ruskin, Koranda and Masino178) ). Improved clearance and levels of protein transporters for neurotransmitters was reported to improve synaptic transmission. Cortical excitability was described as improved due to a reduction in long-term potentiation, without any change in baseline excitability or impact on normal brain activity. Not all studies noted reduced long-term potentiation(Reference Huang, Li and Wu67).
-
14. Vascular supply (reported in three studies across three disease models (Reference Ma, Wang and Parikh48,Reference Yang, Guo and Wang61,Reference Viggiano, Meccariello and Santoro81) ). The size of cerebral infarct and oedema was reduced with a ketogenic diet. Alzheimer’s models reported increased blood flow providing positive outcomes. In epilepsy, positive outcomes due to a decrease in capillarisation associated with seizures were also reported.
Table 1. Overall themes presented for beneficial ketogenic diet outcomes

ARD, age-related degeneration; ALZ, Alzheimer’s disease; AUT, autism; CI, cerebral ischaemia; CP, chronic pain; CNS, central nervous system generally; D, diabetes; EP, epilepsy; MetS, metabolic syndrome; MCI, mild cognitive impairment; MS, multiple sclerosis; NT, nerve toxin; ON, optic nerve; PKD, Parkinson’s disease; PND, peripheral nerve dysfunction; SCI, spinal cord injury; ST, stroke; TBI, traumatic brain injury.
Discussion
The aim of this scoping review was to investigate animal models that report outcomes related to the nervous system by changing from a standard animal diet to a ketogenic diet. We identified fourteen broad themes of biological mechanisms from eighteen different disease models by which a ketogenic diet is reported to influence the nervous system in animal models (Table 1). Multiple themes may be present within a single study, with many of the different mechanisms and pathways reported resulting in similar overarching effects, including reduction of inflammation and oxidative stress, normalisation of neuronal excitability and improved cell viability. The themes outlined are consistent with other broader reviews that included in vitro and hypothetical models(Reference Yang, Shan, Zhu, Wu and Wang211,Reference Morris, Puri and Maes212) . The purpose of describing these themes was to provide insight into how altering dietary macronutrients to produce ketosis in humans could also plausibly exert influence on the nervous system in a chronic pain model. The ketogenic diet appears to utilise metabolic modulation to engage the reported mechanisms in animal studies, and thus could also potentially facilitate positive changes within a human nervous system that has undergone aberrant neuroplasticity leading to a persistent pain state.
There are many mechanisms presented that fit with current priorities in pain neuroscience research, such as targeting inflammation. An increase in pro-inflammatory cytokines is often seen in chronic lifestyle disease(Reference Spite, Clària and Serhan213), but also frequently occurs with chronic pain(Reference Totsch, Waite, Sorge, Theodore and Gregory214,Reference Farrell, de Zoete, Cabot and Sterling215) . The failure of the inflammatory response to resolve perpetuates the development of metabolic diseases, but also potentially contributes to persisting pain by shifting the nervous system towards a pathologically maladapted state(Reference Schistad, Stubhaug, Furberg, Engdahl and Nielsen216). Neuroinflammation is a common finding in many neurological conditions and was frequently reported in the outcomes from the extracted studies. Modulation of neuroinflammation across various models from the ketogenic diet was attributed to as many as nine mechanisms (Supplementary Table 2): (a) suppression of nuclear factor (NF)-kβ resulting in reduced expression of proinflammatory cytokines tumour necrosis factor α (TNF-α), interleukin (IL)-1β and interferon (IFN)-γ(Reference Lu, Yang and Zhou196); (b) a decrease in hippocampal mRNA levels of IL-1β(Reference Dupuis, Curatolo, Benoist and Auvin106); (c) reduced pro-inflammatory cytokine hippocampal TNF-α levels with reduced NF-kβ dependant cyclooxygenase (COX)-2 (enzyme for prostaglandin synthesis) signalling pathway(Reference Jeong, Jeon and Shin123); (d) activation of the peroxisome proliferator-activated receptor (PPAR)-γ(Reference Simeone, Matthews, Samson and Simeone161) (a nuclear transcription factor involved in detecting and metabolising lipids) which also suppresses the COX-2 dependant pathway(Reference Jeong, Jeon and Shin123) and regulates catalyse expression(Reference Knowles, Budney and Deodhar126); (e) central and peripheral suppression of inflammatory cytokines/chemokines coupled with a reduction in reactive oxygen species (ROS)(Reference Kim, Hao and Liu183); (f) meeting the cellular energy demand which inhibits AMP-activated protein kinase (AMPK) (which senses and regulates cellular energy levels) and reduces low-energy facilitated inflammation(Reference Harun-Or-Rashid and Inman188); (g) inhibition of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome through ketone action on hydroxycarboxylic acid receptor 1 (HCAR1)(Reference Harun-Or-Rashid and Inman188,Reference Lu, Yang and Zhou196) ; (h) altered NAD+/NADH ratio (which is coupled to glycolysis) and regulates inflammation(Reference Elamin, Ruskin, Masino and Sacchetti63); and (i) reduced mitochondrial ROS production(Reference Guo, Wang and Zhao199). The use of a ketogenic diet for chronic pain management could be theoretically targeting any of these mechanisms to lower inflammation and reduce pain perception(Reference Ruskin and Masino4,Reference Masino and Ruskin18,Reference Dupuis and Masino217) , and is supported mechanistically by the outcomes from animal research.
Mitochondrial pathology is another theme presented that has been implicated in central sensitisation seen in chronic pain, with dysfunctional mitochondria observed in the muscle cells of fibromyalgia patients(Reference Meeus, Nijs, Hermans, Goubert and Calders218), and two recent studies reporting between 67 % and 91 % of patients with mitochondrial diseases also reporting chronic pain(Reference Löffler, Gamroth, Becker and Flor219,Reference Van Den Ameele, Fuge and Pitceathly220) . Given this, strategies to restore or optimise mitochondrial function would be an appropriate pain management strategy(Reference Sui, Xu and Liu221). Beneficial outcomes on mitochondria were frequently reported in the extracted studies (Table 1); however, the result is less clear when examining the outcomes of individual studies (Supplementary Table 2). Kephart et al.(Reference Kephart, Mumford and Mao180) reported no benefit to mitochondrial quality in brain tissue sampled following a long-term ketogenic diet. A study by Lauritzen et al.(Reference Lauritzen, Hasan-Olive and Regnell45) was one of the few to report negative outcomes. This study was designed specifically to examine a mouse model of mitochondrial dysfunction bred to express a mutated mitochondrial DNA repair gene (mutUNG1) designed to represent DNA damage that occurs in neurological disorders. They reported an increase in mitochondrial mass in the hippocampus and upregulated mitochondrial antioxidant defences, which would appear positive; however, this did not correlate with their overall observation of accelerated neurodegeneration from impaired mitochondrial dynamics and function. The context of their experiment becomes important, where the ketogenic diet increased mitochondrial biogenesis, but this increase was of dysfunctional mitochondria, compounding the neurodegeneration and energy demands. This study highlights the difficulty in extrapolating these results to human application. Their research does not necessarily apply to a ketogenic diet applied in the absence of this specific mitochondrial gene mutation, but as the authors(Reference Lauritzen, Hasan-Olive and Regnell45) conclude, the diet also cannot be considered always beneficial for every type of mitochondrial pathology. Theoretically, the ketogenic diet appears to have potential for pain management through the improvement of mitochondrial function with subsequent reduction of oxidative stress and inflammation. Variability in clinical efficacy is likely to exist due to nuance in the mechanism of mitochondrial pathology.
Difficulty in extrapolating results also exists where an animal is fed the diet, but the analysis occurs in a dissected animal which is no longer a part of a complex adaptive system. One of the inclusion criteria for the current review was that the experiment had to have fed a ketogenic diet to the animal; cell culture and in vitro studies were excluded. The lack of an intact noradrenergic system may limit the effect of the ketogenic diet and produce disparate results(Reference Szot, Weinshenker, Rho, Storey and Schwartzkroin164) and may also account for the differences seen between animal and human trials involving ketogenic diets.
Chronic pain involves an increase in neuronal excitability(Reference Parker, Lewis, Rice and McNair222,Reference Neblett, Cohen and Choi223) , with links suggested between these mechanisms and those involved in seizures, and the use of anticonvulsant medications to treat neuropathic pain(Reference Masino and Ruskin18). A ketogenic diet has been widely used clinically as a treatment for epilepsy with several trials in adults(Reference Mahmoud, Ho-Huang and Buhler15) as well as children(Reference Rezaei, Abdurahman, Saghazadeh, Badv and Mahmoudi224). A similar interpretive difficulty lies in the animal research for epilepsy where clinical human trials report generally favourable outcomes, but the animal research results can range between anticonvulsant to pro-convulsant outcomes(Reference Bough, Matthews and Eagles91,Reference Thavendiranathan, Mendonca and Dell166,Reference Zarnowska, Luszczki and Zarnowski175) (Supplementary Table 2). Again, experiment design becomes important, with the eighty-nine epilepsy studies including: different animal models (species, strain and age), multiple different seizure induction models (using different chemicals with different target receptors, and some using electrical shock), inconsistent levels of ketosis achieved, different chow content and quality, different chow quantity (with some diets employing calorie restriction in conjunction with the ketogenic diet), different lengths of dietary intervention, mismatched animal weight between groups resulting from different diets(Reference Nylen, Likhodii, Abdelmalik, Clarke and Burnham150), and different dietary applications where the diet could be started pre-seizure/brain injury or after the event. Despite commonalities, translating the proposed neuromodulatory mechanisms from the animal epilepsy research to clinical chronic pain conditions requires more nuance and may explain variable clinical results in any human trials.
Neurotransmitter function was frequently reported in the included studies as a change within the nervous system favouring a reduction or restoration of normal levels of neuronal excitability. The mechanism reported was improved GABA-to-glutamate ratios usually via increased GABA (inhibitory) and/or decreased glutamate (excitatory) levels, with outcomes being a reduction in various seizure metrics in the animals tested. The research exploring the relationship between chronic pain and neurotransmitter levels is inconsistent. There is evidence supporting motor cortex disinhibition that is more pronounced in neuropathic pain(Reference Parker, Lewis, Rice and McNair222); however, whether this is due to a loss of GABAergic inhibition, as has been suggested, is still unclear. A recent systematic review reported altered neurotransmitter levels demonstrated in a small number of human chronic pain trials. There were increased levels of Glx (glutamate and glutamine combined) reported, but no corresponding reduction in GABA as might be expected(Reference Peek, Rebbeck and Puts225). The authors reported that different pain conditions may present with unique neurometabolite signatures, but the research was limited by inadequate reporting and standardisation of magnetic resonance spectroscopy techniques used.
A further variable that may contribute to the inconsistencies reported is that of the chow. Problems exist where the control diets are not matched appropriately to the ketogenic chow. Differences in vitamins, minerals and fibre exist between the diets as well as the macronutrient properties, limiting the ability to assess the ketogenic component of the diet. A number of issues also exist with the commercial rodent ketogenic diet formulations, including restriction of protein, choline deficiency(Reference Schugar, Huang, Moll, Brunt and Crawford226) and poor-quality fats (such as hydrogenated vegetable oils) rather than fats with a more beneficial inflammatory profile (such as omega-3)(Reference Anez-Bustillos, Dao and Finkelstein227).
The evidence presented in animal models supporting positive changes from a ketogenic diet, such as seen with anti-inflammatory mechanisms, appears compelling. However, the reported outcomes overall are often inconsistent and ambiguous(Reference Huang, Li and Wu67), and there are many difficulties when extrapolating from animal models to human models of chronic pain(Reference Burma, Leduc-Pessah, Fan and Trang228). The use of specific animal strains and sex may reduce the heterogeneity and increase the likelihood of detecting an effect, but may be poor representations of the diversity in target human pain populations(Reference Klinck, Mogil and Moreau229). These translational issues could be explored by also including natural animal models (such as using the ketogenic diet on naturally occurring pain presentations in domestic animals)(Reference Klinck, Mogil and Moreau229) as well as more consistency in experimental design, and reporting which more clearly acknowledges the limitations of the research. These strategies may allow the data to better inform human clinical trials of chronic pain.
Conclusion
Fourteen broad themes were identified from the literature outlining how a ketogenic diet influences nervous system function from animal models. The mechanisms presented centred around the reduction of inflammation and oxidative stress as well as a reduction in nervous system excitability. These mechanisms are potential drivers of chronic pain, and treatment strategies which target these have implications for chronic pain management. Given the multiple potential mechanisms presented, it is likely that many of these are involved synergistically and undergo adaptive processes within the human body, and controlled animal models that limit the investigation to a particular pathway in isolation may reach differing conclusions. Attention is required when translating this information to human chronic pain populations owing to the limitations outlined from the animal research.
Financial support
This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sector.
Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0954422421000214