


The earliest link between cholesterol deposition in arterial walls and death from coronary heart disease (CHD) was documented by Virchow over a century ago (Ref. Reference Steinberg and Gotto1), but it was not until the 1950s that prospective examination of the relationship between levels of plasma LDL (low-density lipoprotein)-C (cholesterol) and CHD was reported by the Framingham Study (Ref. Reference Kannel and McGee2). It is beyond doubt that the two are intrinsically linked, and there is a direct correlation between the levels of plasma LDL-C and the risk of CHD (Ref. Reference Grundy3). The elucidation of the cholesterol synthetic pathway has facilitated the development of therapeutic strategies for lipid lowering and, as a result, hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) first came into clinical use during the 1980s. Statins inhibit the rate-limiting step in intracellular cholesterol synthesis, that of the conversion of HMG-CoA to l-mevalonate, by binding to the active site of HMG-CoA reductase (Ref. Reference Istvan and Deisenhofer4). This effectively decreases cholesterol synthesis by the liver, leading to upregulated LDL receptor expression, with consequent increased clearance of cholesterol from the plasma.
The first statins used clinically were lovastatin and simvastatin, the latter being evaluated in a landmark trial – the Scandinavian Simvastatin Survival Study (4S) (Ref. 5). This study revealed an unequivocal reduction in all-cause mortality in 4444 hypercholesterolaemic patients with CHD who were followed up for 5 years, and provided the first definitive evidence of improved survival with statin therapy. Over the past two decades, statin therapy has become widely prescribed, and these drugs are probably the most thoroughly investigated primary therapeutic agents in clinical trials (Ref. Reference Brugts6).
Statins in clinical trials
In the 4S study, the ability of statins to reduce mortality in individuals with a history of CHD was considered to be attributable entirely to their cholesterol-lowering properties. These conclusions were supported by two further large studies, CARE (Ref. Reference Sacks7) and LIPID (Ref. 8), that evaluated the outcomes of cholesterol lowering in approximately 18 000 patients treated with pravastatin. These two studies also described a dramatic and consistent effect in patients with pre-existing CHD but without elevated cholesterol. A subsequent study of more than 20 000 patients in the UK provided significant new knowledge in this respect. The seminal findings of the Heart Protection Study (HPS) concluded that substantial benefit from statin therapy was apparent in individuals not only with existing CHD, but also with diabetes, cerebrovascular disease and peripheral arterial disease (Ref. 9). The study clearly showed that simvastatin therapy was equally beneficial to individuals with high and ‘normal’ levels of cholesterol and therefore raised the question of whether these benefits could be entirely attributable to effects on cholesterol alone.
A large number of subsequent clinical trials have concluded beyond doubt that statin therapy has proven effectiveness in decreasing CHD and reducing cardiovascular mortality. Subgroup analysis of some of these previous trials demonstrated that effects beyond lipid lowering were apparent. In the WOSCOPS (Ref. Reference Shepherd10) and CARE (Ref. Reference Sacks7) trials, risk of CHD was significantly reduced in statin-treated age- and sex-matched individuals with similar absolute levels of cholesterol. In 2005, the Cholesterol Treatment Trialists (CTT) Collaborators (Ref. Reference Baigent11) performed a meta-analysis of 14 randomised studies involving over 90 000 participants followed for 5 years. These collated data clearly demonstrated that statin therapy reduced the incidence of major coronary events, coronary revascularisation and stroke, and was related to absolute reduction in cholesterol, irrespective of initial levels. It is believed that half of all cases of myocardial infarction (MI) and stroke occur in apparently healthy individuals with plasma cholesterol levels below the threshold for lipid-lowering treatment (Ref. Reference Ridker12), suggesting that other contributing factors have a role. The JUPITER trial was a large randomised study conducted in 26 countries, recruiting almost 18 000 apparently healthy men and women with no history of cardiovascular disease. Although these individuals had ‘normal’ cholesterol levels, they exhibited increased levels of C-reactive protein (CRP), an inflammatory biomarker proposed to be predictive of future vascular events. Administration of rosuvastatin significantly reduced the rate of primary cardiovascular events in these patients (Ref. Reference Ridker12), the results being so clear-cut (major cardiovascular events in rosuvastatin group 142, versus 251 in placebo) that the trial was terminated after a median follow-up of 1.9 years (maximal 5 years).
The unequivocal outcomes of several clinical studies supported the widespread prescribing of statins to individuals at risk, such that some investigations have adopted more aggressive approaches to lipid lowering. Several studies, including PROVE-IT (Ref. Reference Cannon13), TNT (Ref. Reference LaRosa14), IDEAL (Ref. Reference Pedersen15), SEARCH (Ref. Reference Armitage16) and A to Z (Ref. Reference de Lemos17), have reported that high-dose statin therapy can further reduce cardiovascular risk compared with standard regimens. These data from a total of almost 40 000 patients are collated in the most recent meta-analysis of 26 statin trials by the CTT Collaborators, comparing intensive versus regular statin treatment (Ref. Reference Baigent18).
Clinical evidence for statin pleiotropy
Pleiotropy refers to actions of a drug that extend beyond that for which it was specifically developed. Such effects are generally unexpected and can be related to the drug's primary mode of action or may possibly be unrelated (Ref. Reference Davignon19). It has traditionally been assumed that the key benefits of statin therapy are attributable to the lowering of serum cholesterol levels, but the concept of pleiotropism first arose through retrospective analysis of some early clinical trials. In particular, in the 4S and TNT trials, a reduction in the incidence, morbidity and mortality of heart failure was reported (Refs Reference LaRosa14, Reference Kjekshus20). In another clinical study, statin treatment for 14 weeks led to improved exercise endurance that corresponded to a significant increase in left ventricular (LV) ejection fraction and a decrease in plasma inflammatory cytokines: effects not observed in the placebo control group (Ref. Reference Node21).
Further support for the notion that additional mechanisms of action exist with statin therapy is that the risk of MI in statin-treated patients is lower than that of patients receiving alternative cholesterol-lowering strategies (Refs Reference Corsini22, Reference Liao and Laufs23, Reference Pekkanen24). For example, ezetimibe, a drug that lowers cholesterol by impeding intestinal absorption, does not offer equivalent cardioprotective effects to statins despite comparable reductions in serum cholesterol (Refs Reference Liu25, Reference Ostad26). In the JUPITER trial, the significance of inflammatory mediators as a marker of future cardiovascular events was highlighted and the measured reduction in CRP following statin therapy was unrelated to a reduction in cholesterol levels. Moreover, the patients enrolled in that study had no history of cardiovascular disease and had cholesterol levels in the normal range (Ref. Reference Ridker12). In summary, accumulating evidence supports the existence of statin pleiotropy in the cardiovascular system. To what extent these effects justify their clinical benefits in the heart is less clear-cut but will now be reviewed in the light of current data.
Statins and the heart
The beneficial impact of statin therapy on cholesterol reduction is beyond doubt, and recognition of its far-reaching effects beyond lipid lowering is now widely documented. These properties include restoration of aberrant endothelial function, reducing oxidative stress, stabilisation of atherosclerotic plaques, inhibition of platelet aggregation, inhibition of vascular smooth muscle proliferation and migration, and reducing vascular inflammation (reviewed in Ref. Reference Liao and Laufs23).
Although most knowledge has been acquired through studies in vascular tissue and cells, data from clinical studies have provided evidence that statins also exert direct effects on the myocardium. In the Western world, congestive heart failure is a key cause of morbidity and mortality, and there are now several lines of evidence supporting the use of statins in heart failure patients. Indeed, it was retrospective analysis of the 4S study which showed that patients on long-term simvastatin therapy had reduced incidence of heart failure (Ref. Reference Kjekshus20). Statins reduce LV mass in hypertensive patients (Ref. Reference Su27) and lower the incidence of atrial fibrillation in patients with coronary artery disease (Ref. Reference Young-Xu28). Most large statin trials have subsequently documented favourable outcomes in heart failure patients (reviewed in Ref. Reference Tsouli29), perhaps unsurprisingly, owing to the underlying commonality of atherosclerotic disease in ischaemic heart failure (Ref. Reference LaRosa14). However, statins are also beneficial in patients with heart failure of non-ischaemic aetiology (Refs Reference Node21, Reference Foody30, Reference Horwich, MacLellan and Fonarow31). Such observations support a mechanism that is independent of cholesterol and are reportedly additional to effects achieved with standard heart failure therapies such as ACE inhibitors and β-blockers (Ref. Reference Ramasubbu32).
The consensus view of clinical studies with statins in patients with heart failure is the exhibition of several clinical and biological effects with a diversity of reported mechanisms: anti-inflammatory, antioxidant, improvements in endothelial function, increased angiogenesis, reduced cardiac hypertrophy and remodelling, reduced neurohormonal activation, improved autonomic regulation and anti-arrhythmic effects (reviewed in Ref. Reference Ramasubbu32). However, whereas small prospective studies and post-hoc analyses of randomised trials support the notion that statins are effective in patients with heart failure, the recent randomised controlled GISSI-HF study specifically addressed their use in patients with heart failure. In this prospective trial of more than 4600 patients, it was reported that the standard 10 mg administration of rosuvastatin had no beneficial effect, irrespective of heart failure aetiology (Ref. Reference Tavazzi33). It is perhaps pertinent to note that the broad population of patients recruited to this study had established, symptomatic heart failure and were receiving traditional heart failure therapies. Therefore, it is conceivable that addition of statin therapy is less effective in reducing cardiovascular events when heart failure is established, as opposed to prevention of heart failure in a population with CAD. This suggestion is endorsed by studies in which different statin regimens led to a consistent reduction in the development of heart failure in CAD patients (Refs Reference de Lemos17, Reference Kjekshus20, Reference Khush34, Reference Scirica35). Some studies propose that statin therapy may be beneficial early after acute ischaemic events that are associated with LV dysfunction and failure (Ref. Reference Kumar and Cannon36). In further support of this proposition, statins do not appear to improve outcomes in patients with advanced heart failure (Ref. Reference Laufs, Custodis and Bohm37). The use of statins as an additional preventive strategy for patients at risk of atrial fibrillation does however appear to be beneficial (reviewed in Ref. Reference Adam38).
Outside the clinical scenario, the weight of evidence from animal studies clearly supports the concept that statins confer beneficial effects on adverse myocardial remodelling. These include improvements in LV remodelling, hypertrophy and fibrosis, with resultant enhanced cardiac function (Refs Reference Bauersachs39, Reference Hasegawa40, Reference Hayashidani41, Reference Luo42, Reference Patel43). Regardless of the initiating stimulus (aortic banding, MI or a variety of transgenic models), the benefits of statin administration were clear. These studies were conducted in normocholesterolaemic animals with no induction of atherosclerosis; therefore, the documented effects were independent of correcting hyperlipidaemia. Rather, the mechanism of the effects of statins appears to be due to inhibition of small GTPases, which are important modulators of myocardial function.
Evidence for small GTPases in mediating myocardial remodelling
The multiplicity of reported pleiotropic effects of statins appears to be attributable to their ability to inhibit synthesis of intracellular mevalonate: a key upstream precursor of not only cholesterol, but also important isoprenoid intermediates, farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) (Ref. Reference Liao44) (Fig. 1). These intermediates have key roles in post-translational modification of a range of proteins, including small GTPases (Ras, Rho, Rac), by serving as lipid attachments through a process known as prenylation. In this respect, isoprenoid moieties permit attachment, subcellular localisation and trafficking of membrane-bound proteins coupled to intracellular signalling pathways, thus facilitating changes in cellular function (Ref. Reference Van Aelst and Souza-Schorey45). The key role of prenylated proteins in the control of cellular function significantly affects cellular homeostatic activity in which most processes are regulated, either directly or indirectly, by Rho or Ras GTPases (Ref. Reference Lezoualc'h46). In addition, Rac1 GTPase contributes to myocardial pathologies both in experimental studies (Refs Reference Adam47, Reference Custodis48) and in humans (Ref. Reference Maack49). Hence it is likely, through modulation of such mechanisms, that statins confer beneficial pleiotropic effects. Consistent with this proposal, many of the inhibitory effects of statins in vitro are reversible by isoprenoid (FPP/GGPP) supplementation, but not by cholesterol itself, or its precursor squalene (Fig. 1).

Figure 1. The cholesterol synthesis pathway. Statins inhibit HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis. Resultant inhibition of FPP and GGPP synthesis inhibits prenylation of small GTPases of the Ras and Rho (Rho/Rac/cdc42) families, respectively, resulting in pleiotropic effects that are independent of effects on cholesterol synthesis. Abbreviations: AA-CoA, acetoacetyl coenzyme A; HMG-CoA, hydroxymethylglutaryl coenzyme A; IPP, isopentenyl pyrophosphate; GPP, geranyl pyrophosphate; FPP, farnesyl pyrophosphate; GGPP geranylgeranyl pyrophosphate.
It is now well established that small GTPases have important roles in regulating myocardial remodelling. Studies performed over a decade ago first demonstrated the importance of small GTPases, particularly Ras and Rho family proteins, in signalling from G-protein-coupled receptors to development of myocardial hypertrophy (reviewed in Refs Reference Clerk and Sugden50, Reference Hunter and Chien51). Interestingly, Ras, Rho and Rac have all been implicated in promoting cardiac hypertrophy (Ref. Reference Brown, Del Re and Sussman52). For example, typical hypertrophic agents such as phenylephrine and endothelin-1 (ET-1) have been shown to activate Rac1 through specific intracellular signalling pathways in cardiac myocytes (Ref. Reference Clerk53). In another study, cardiac-specific deletion of Rac1 in a murine model reduced angiotensin II (Ang II)-mediated hypertrophy (Ref. Reference Satoh54); conversely, cardiac-specific Rac1 overexpression led to atrial fibrillation (Ref. Reference Adam47). Interestingly, increased Rac1 activity has been associated with cardiac hypertrophy in humans (Ref. Reference Lu55).
Transgenic mice with cardiac-specific expression of RhoA, whilst showing no obvious hypertrophy, had clear evidence of heart failure accompanied by oedema, ventricular and atrial dilation, and increased fibrosis (Ref. Reference Sah56). Rho kinase (ROCK) is a well-characterised downstream effector of RhoA, and studies in which ROCK was inhibited have indicated a role in diverse cardiovascular pathologies. Pharmacological inhibition of ROCK implicates it in the development of cardiac hypertrophy in rats (Refs Reference Higashi57, Reference Satoh58) and mice (Ref. Reference Wang59). In murine studies, long-term inhibition of ROCK suppressed LV remodelling after MI (Ref. Reference Hattori60) and protected from ischaemia-reperfusion injury (Ref. Reference Bao61). In transgenic models, ROCK1 haploinsufficient mice showed decreased cardiac fibrosis after MI (Ref. Reference Rikitake62), and targeted deletion of ROCK significantly reduced fibrosis after three weeks of pressure overload (Ref. Reference Zhang63).
The in vivo function of Ras was first studied in transgenic mouse models (Ref. Reference Hunter64) in which animals displayed characteristic hypertrophy with features in common with human disease (Ref. Reference Zheng65). Indeed, a correlation between Ras expression and the severity of hypertrophy has been reported in humans (Ref. Reference Kai66).
Given the established roles of small GTPases in the aetiology of heart failure and other cardiac pathologies, inhibition of their activity by statins almost certainly underlies many of the pleiotropic effects of these drugs.
Effects of statins on resident myocardial cells
Evidence collected from animal models indicates that statins can ameliorate adverse myocardial remodelling largely through effects on Ras, Rho and Rac, but their precise site of action and specific cellular effects are difficult to elucidate in this setting. It is established that activation of small GTPase signalling in the heart is detrimental to cardiac function, but what is less clear is how blocking these pathways leads to beneficial effects at the cellular level. In the mammalian heart, cardiac function is dynamically regulated by the two key resident cell types, cardiomyocytes and cardiac fibroblasts (CFs). The normal human heart is composed of 30% cardiomyocytes and 70% nonmyocytes, the majority of which are CFs (Ref. Reference Jugdutt67). CFs are primarily responsible for extracellular matrix (ECM) homeostasis and regulate the structure of the heart, thereby providing coordinated signals between cellular and noncellular components (reviewed in Ref. Reference Porter and Turner68). Cardiomyocytes, although fewer in number, occupy the bulk volume and provide mechanical force, transmission of which is a key function of the ECM. It is acknowledged that statins can also modulate other nonmyocyte cells resident in the heart, including endothelial cells, smooth muscle cells, neuronal cells and mast cells. However, these have been recently reviewed elsewhere (Ref. Reference Zhou and Liao69) and will not be discussed further here.
Historically, cardiac myocytes have been the key focus of clinical and experimental studies in vivo and in vitro. Although CFs have received considerably less attention, their role in regulating and maintaining the structural integrity of the heart through controlled proliferation and ECM turnover is pivotal to cardiac function (Refs Reference Porter and Turner68, Reference Brown70, Reference Camelliti, Borg and Kohl71, Reference Weber72). CFs respond to a variety of stimuli (mechanical, electrical and chemical) to coordinate cell–cell and cell–matrix interactions in the maintenance of cardiac homeostasis (Refs Reference Banerjee73, Reference Kohl74). CFs regulate ECM turnover by balancing the synthesis of ECM components (collagens, laminins, glycosaminoglycans, matricellular proteins) with their breakdown, through the production of an array of specific proteases, including members of the matrix metalloproteinase (MMP) family (Ref. Reference Porter and Turner68). Although electrically unexcitable, CFs are coupled to myocytes and can influence myocyte electrophysiology (Ref. Reference Camelliti, Green and Kohl75).
The heart undergoes adaptive remodelling in response to pathophysiological stresses, including hypertension-associated pressure overload and ischaemia-reperfusion injury after MI: changes that in the longer term can progress to pathological remodelling and heart failure. At the level of the cardiomyocyte, these adaptive changes include loss of contractile function (through changes in contractile protein expression, excitation–contraction coupling and desensitisation to β-adrenergic signalling), increased cell hypertrophy, and augmented apoptotic and necrotic cell death (Ref. Reference Mann and Bristow76). Concurrently, CFs undergo phenotypic transformation to become myofibroblasts, display an increased propensity for cell proliferation and migration, and modulate ECM turnover through elevated MMP expression and enhanced collagen deposition (fibrosis) (Ref. Reference Porter and Turner68). Therefore, complex alterations in both myocyte and fibroblast biology occur during adaptive and pathological myocardial remodelling.
Given this heterogeneity, it is clear that multiple cellular and molecular mechanisms underlie the benefits of statins on myocardial remodelling. Investigation of the modulatory effects of statins on individual myocardial cell types in vitro has therefore shed more light on their cellular and molecular mechanisms, as reviewed below.
Effects of statins on cardiac myocytes
In vivo and in vitro studies have provided evidence that statins can reduce the detrimental effects of myocardial injury on cardiomyocyte function at multiple levels, including hypertrophy, cell death and contractile function (Fig. 2a). The use of in vitro cell cultures in particular has enabled delineation of intracellular mechanisms of action of statins on these aspects of myocyte function. It should be noted, however, that the vast majority of these in vitro studies have utilised cultures of neonatal cardiomyocytes as model systems rather than mature differentiated adult cardiomyocytes, so an element of caution is warranted in translating these findings to the adult in vivo system, particularly that of man.

Figure 2. Effect of statins on cardiomyocyte and cardiac fibroblast function. Summary of some of the most well-established effects of statins on (a) cardiomyocyte and (b) cardiac fibroblast (CF) function. Statins can reduce cardiomyocyte hypertrophy and cell death, and improve electrical and contractile function following myocardial injury. Statins can also attenuate CF proliferation, migration, myofibroblast differentiation and extracellular matrix (ECM) synthesis.
Hypertrophy
Statins are widely accepted to have beneficial effects by reducing cardiac hypertrophy in experimental animal models (Ref. Reference Mital and Liao77). In vitro studies on the effects of statins on markers of neonatal cardiomyocyte hypertrophy have also shown that the stimulatory effects of hypertrophic agonists (e.g. Ang II, ET-1, NE) are opposed by lipophilic statins, but not by the hydrophilic pravastatin which appears to have no effect in vitro (Refs Reference Oi78, Reference Nishikimi79). The underlying mechanisms by which inhibition of HMG-CoA reductase modulates the hypertrophic response are mostly cholesterol independent, involving inhibition of GTPase prenylation (summarised in Fig. 3), particularly those of the Rho and Rac family that are integral to the hypertrophic mechanism (Ref. Reference Brown, Del Re and Sussman52). By preventing geranylgeranylation and membrane association of RhoA, statins prevent downstream signalling to Rho substrates including ROCK, which is an important regulator of the actin cytoskeleton and cell size (Refs Reference Nishikimi79, Reference Laufs80, Reference Morikawa-Futamatsu81). The small GTPase Rac1 is crucial for activation of NADPH oxidase, a mediator of Ang II and ROS-mediated myocyte hypertrophy (Ref. Reference Hordijk82). By reducing Rac1 geranylgeranylation, statins attenuate NADPH oxidase activity and subsequent Ang-II-induced myocyte hypertrophy (Refs Reference Custodis48, Reference Laufs80, Reference Takemoto83). The ability of statins to inhibit farnesylation of the small GTPase p21Ras, and thus downstream ERK signalling, might also contribute to statin-mediated reductions in myocyte hypertrophy (Ref. Reference Oi78). More recent studies have focused on the potential effects of statins on additional prenylated proteins, including large G-proteins that may modulate cellular responses to hypertrophic stimuli. For example, rosuvastatin was shown to inhibit α1-adrenergic-receptor-induced cardiomyocyte hypertrophy by suppressing expression and membrane association of the large G-protein G h, resulting in reduced PKC–ERK signalling (Ref. Reference Choi84). This is significant because it has been shown that coupling of the α1-receptor to G h can aggravate cardiac remodelling in the failing human heart (Ref. Reference Hwang85).
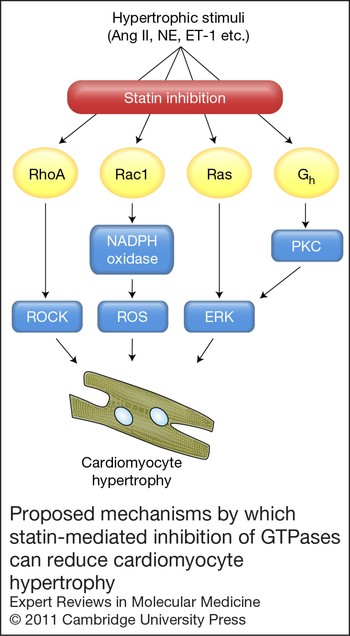
Figure 3. Proposed mechanisms by which statin-mediated inhibition of GTPases can reduce cardiomyocyte hypertrophy. Statins inhibit prenylation of small GTPases of the Ras, Rho and Rac families, resulting in reduced cardiomyocyte hypertrophy. Statins may also modulate large G-protein function (e.g. Gh) to similar effect.
Additional mechanisms of action of statins on hypertrophic signalling pathways have also been reported, which appear to be initiated independently of effects on G-proteins, including direct modulation of the JAK–STAT (Refs Reference Wu86, Reference Liu, Shen and Wu87) and PI3K–AKT (Refs Reference Planavila88, Reference Hauck89) pathways.
Cell death
Therapies aimed at reducing cardiomyocyte cell death hold promise in reducing infarct size and improving clinical outcomes (Refs Reference Whelan, Kaplinskiy and Kitsis90, Reference Webster91). Statins are reported to exert cardioprotective effects by reducing myocyte cell injury and death (Ref. Reference Webster91). In vitro studies have investigated the underlying mechanisms at the level of the cardiomyocyte and current thinking is that statins activate the PI3K–AKT pathway (Refs Reference Verma92, Reference Bergmann93) and other components of the reperfusion injury salvage kinase (RISK) pathway (Ref. Reference Vilahur94), leading to increased NO synthesis (Refs Reference Verma92, Reference Jones95) and reduced mitochondrial dysfunction (Ref. Reference Jones95). Rac1 might also be a target for the antiapoptotic effects of statins, as cerivastatin reduced β-adrenergic-receptor-induced apoptosis and Rac1 and JNK activity in adult rat cardiomyocytes (Ref. Reference Ito96).
Despite these studies, the impact of statins on cardiomyocyte apoptosis is not as clear-cut as it is for myocyte hypertrophy, and several other studies have suggested that rather than reducing apoptosis, statins may actually augment myocyte death. For example, simvastatin promoted apoptosis of adult human cardiomyocytes, effects that were prevented by co-supplementation with mevalonate or GGPP, indicative of a cholesterol-independent mechanism involving inhibition of protein geranylgeranylation (Ref. Reference Demyanets97). Similarly, in neonatal rat cardiomyocytes, fluvastatin induced apoptosis that was reversed by mevalonate or GGPP and was mimicked by a protein prenylation inhibitor (perillic acid) or the ROCK inhibitor Y27632 (Ref. Reference Ogata98). It should be noted, however, that cardiomyocytes (especially those from adult hearts) are particularly difficult to maintain in culture, partly because the ECM transduces key survival signals in vivo that cannot be simulated in vitro (Ref. Reference Rivera and Lowes99). One should therefore be tentative in applying knowledge from in vitro apoptosis experiments to the in vivo scenario.
Electrical and contractile function
Statins may improve cardiac contractile function by increasing expression of sarcoplasmic calcium regulatory proteins, including SERCA and RyR2 (Refs Reference Choi84, Reference Zheng and Hu100). Statins can also modulate myocyte action potentials through indirect effects on Kv1.5 and Kv4.3 channel activity (Ref. Reference Vaquero101), observations that might underlie the reported antiarrhythmic effects of statins both in experimental models and in humans (Ref. Reference Lee, Blaha and Jones102). A further beneficial effect of statins might be in opposing the potentially harmful positive inotropic effects of β-adrenergic stimulation. Atorvastatin can desensitise cardiomyocytes to the effects of β-adrenergic stimulation through a mechanism involving reduced isoprenylation of γ-subunits of heterotrimeric G-proteins and subsequent reductions in the cellular content of Gαs and hence cyclic AMP levels (Ref. Reference Muhlhauser103). Similar effects have been observed in rat hearts in vivo (Ref. Reference Schmechel104). Together, these data suggest that the ability of statins to improve cardiac function may be due, at least in part, to direct pleiotropic effects on cardiomyocyte calcium handling and excitation–contraction coupling.
Effects of statins on cardiac fibroblasts
The critical role of CFs in maintaining ECM homeostasis in the healthy heart is underscored by the need to provide structure, function and connectivity for all the myocardial cell types (Ref. Reference Souders, Bowers and Baudino105). Through cell–cell interaction and secretion of growth factors, myocyte function is directly modulated by CFs (Ref. Reference Baudino106). During the myocardial remodelling that follows MI and during heart failure progression, CFs undergo activation to a myofibroblast phenotype, expressing α-smooth muscle actin (α-SMA) and exhibiting proliferative, migratory and secretory properties (Refs Reference Porter and Turner68, Reference Camelliti, Borg and Kohl71). Myofibroblasts can develop not only from pre-existing resident myocardial fibroblasts, but also from endothelial- and epithelial-to-mesenchymal transition, bone marrow stem cells and monocytes (Refs Reference Krenning, Zeisberg and Kalluri107, Reference Zeisberg and Kalluri108). Myofibroblasts are not found in healthy myocardium, but are abundant in granulation tissue in infarct zones, where they have been observed to persist for months (Refs Reference Sun and Weber109, Reference Willems110), or years (Ref. Reference Jugdutt67), in mature scars. It is likely that this persistence facilitates fibrosis that directly influences pathological remodelling and compromises cardiac function that ultimately leads to heart failure.
As outlined in the preceding sections, experimental studies exploring the effects of statins on the heart have mainly focused on global myocardial remodelling in vivo or on isolated cardiomyocytes in vitro to provide evidence that targeting small G-proteins in the heart is beneficial. There are, however, considerably less data relating to the functional effects of statins at the level of CFs. The complexity of the in vivo scenario makes assessment of the direct effects of pharmacological agents on individual cell types difficult. Thus, studies on cultured CFs derived from a variety of sources (animal and human, adult and neonatal, atrial and ventricular) have been the predominant source of current knowledge. With the caveats that CF characterisation is paramount to cellular studies and the relevance of findings to the clinical scenario is interpreted with caution, experimental studies on the effects of statins have revealed important data with respect to myofibroblast differentiation, cell proliferation and migration, and ECM turnover (Fig. 2b).
Myofibroblast differentiation
Although in vivo evidence is lacking, it has been suggested that statins can reduce fibroblast–myofibroblast transformation. For example, simvastatin attenuated expression of α-SMA induced by transforming growth factor-β (TGF-β) in cultured canine atrial fibroblasts (Ref. Reference Shiroshita-Takeshita111) and similarly, pravastatin suppressed phenotypic transformation of rat CFs to myofibroblasts (Ref. Reference Moiseeva112). As ROCK has a key role in differentiation of monocytes to cardiac myofibroblasts (Ref. Reference Haudek113), it is plausible that statins would interfere with this process in vivo; however, this has yet to be tested. Other evidence can be drawn from studies on fibroblasts from noncardiac sources. For example, inhibition of Rac (Ref. Reference Xu114) or ROCK (Ref. Reference Akhmetshina115) prevents human dermal fibroblasts adopting a myofibroblast phenotype, and statins can reduce myofibroblast differentiation (Ref. Reference Meyer-Ter-Vehn116) and epithelial–mesenchymal transition (Ref. Reference Rodrigues-Diez117) by inhibiting RhoA signalling in fibroblasts of non-cardiac origin.
Proliferation and migration
Proliferation and migration of CFs are fundamental to the myocardial-remodelling process, and these functions have been well studied in cultured CFs. A range of statins consistently inhibit CF proliferation regardless of species or mitogenic stimulus. For example, DNA synthesis (a marker of proliferation) in rat (Refs Reference He118, Reference Martin119, Reference Tian120, Reference Xu121, Reference Tian122), mouse (Ref. Reference Chen and Mehta123) and canine (Ref. Reference Shiroshita-Takeshita111) CFs was reduced by statin treatment. Few of these studies have investigated the underlying mechanism for these effects, although inhibition of ERK and AKT signalling pathways by statins has been proposed (Ref. Reference He118). Our own studies using human CFs demonstrated that simvastatin reduced cell proliferation and cyclin A expression in a concentration-dependent manner through inhibition of RhoA geranylgeranylation (Ref. Reference Porter124). Moreover, we further demonstrated in human CFs that simvastatin could inhibit TNF-α (tumour necrosis factor-α)-induced proliferation, migration and invasion (Refs Reference Porter125, Reference Turner126) by distinct mechanisms. Simvastatin reduced the ability of CFs to invade an ECM barrier by reducing secretion of the matrix metalloprotease MMP-9 through a novel post-transcriptional mechanism (Ref. Reference Turner126). Additionally, simvastatin reduced CF migration (motility) per se by inducing cytoskeletal disruption through ROCK inhibition (Ref. Reference Turner126). The known intracellular mechanisms by which statins can reduce CF proliferation and migration are summarised in Figure 4.

Figure 4. Proposed mechanisms by which statins can modulate cardiac fibroblast function. Statins reduce CF migration, proliferation and ECM synthesis by inhibition of RhoA and Rac1 small GTPases. Statins can also inhibit ERK, AKT and p38 MAP kinase signalling pathways, albeit through undefined mechanisms, leading to reduced fibroblast proliferation and collagen synthesis. Additionally, MMP-9 secretion by CFs can be inhibited by statins, thus impeding cell migration through the ECM.
Regulation of ECM
CFs are key regulators of ECM metabolism, responding to altered levels of a range of growth factors and cytokines to balance synthesis and degradation of ECM components (Ref. Reference Jugdutt127). Statins have been consistently found to attenuate collagen synthesis by CFs. For example, procollagen expression was mildly inhibited by pravastatin in Ang-II-treated mouse CFs, an effect that was augmented by co-treatment with pioglitazone, an insulin-sensitising agent (Ref. Reference Chen and Mehta123). The underlying mechanism for this appeared to be the result of reduced Ang-II-induced activation of p38 MAPK and ERK. In neonatal rat CFs, Ang-II- or TGF-β-induced expression of procollagen mRNA and collagen deposition were inhibited by atorvastatin in a concentration-dependent manner (Ref. Reference Martin119). The same study also reported similar effects on human CFs. In addition to directly attenuating collagen synthesis, statins might also exert antifibrotic effects by reducing the secretion of profibrotic growth factors, such as TGF-β or connective tissue growth factor (CTGF). Ang II induces expression of CTGF by activation of Rac1 and NADPH oxidase, and this can be reduced by simvastatin in rat CFs through inhibition of Rac1 (Refs Reference Martin119, Reference Adam128). Atorvastatin can also reduce TGF-β-induced collagen synthesis by inhibiting the PI3K–AKT–Smad3 pathway and reducing expression of endoglin, a membrane glycoprotein that acts as a co-receptor for TGF-β (Ref. Reference Shyu129). Thus, there are multiple mechanisms by which statins can reduce ECM synthesis at the level of the CF (summarised in Fig. 4).
Although CFs derived from several species, including humans, have been shown to express several members of the MMP family (Ref. Reference Porter and Turner68), modulation of this group of proteases by statins is less well studied. Ang II upregulated MMP-3 and MMP-9 expression in mouse CFs and although MMP activity was inhibited by pravastatin, this was only observed when in combination with pioglitazone (Ref. Reference Chen and Mehta123). In our own laboratory, we showed that simvastatin inhibited TNF-α-induced MMP-9 secretion from human CFs and this led to reduced invasive capacity (Ref. Reference Porter125). Studies in other cell types (e.g. vascular smooth muscle and inflammatory cells) have identified MMPs as being potential targets of the pleiotropic effects of statin inhibition (Refs Reference Porter and Turner130, Reference Luan, Chase and Newby131), so it is likely that similar mechanisms exist in CFs.
Anti-inflammatory effects of statins
In statin-treated heart failure patients, reduced plasma levels of proinflammatory cytokines are observed (Refs Reference Tousoulis132, Reference Sola133). Moreover, both experimental and clinical studies suggest that statins can reduce local expression of proinflammatory cytokines in the myocardium (Refs Reference Zhang134, Reference Wallace135). Because CFs are an important source of myocardial cytokines, in our own studies we investigated whether statins could modulate proinflammatory cytokine expression in human CFs (Ref. Reference Turner136). TNF-α treatment stimulated expression of interleukins IL-1α, IL-1β and IL-6, but none was modulated by statin therapy, indicating that CFs are probably not the cellular targets for the anti-inflammatory effects of statins on the heart.
However, we found that exposure of human endothelial cells to inflammatory mediators increased the expression of adhesion molecules and augmented leukocyte adhesions in a flow model (Ref. Reference Eccles137), both of which were abrogated by statin treatment. It appears therefore that the anti-inflammatory effects of statins in the inflammatory milieu of atherosclerosis, CHD and MI are due predominantly to effects on endothelial and inflammatory cells (reviewed in Refs Reference Zhou and Liao69, Reference Arnaud, Braunersreuther and Mach138).
In summary, a large body of evidence supports the concept of both cardiomyocytes and CFs as distinct targets for the beneficial modulatory effects of statins on myocardial remodelling. The ability of statins to regulate cellular function at different levels underscores their utility as versatile therapeutic agents through their pleiotropic effects in addition to their primary role as cholesterol-lowering drugs.
Future directions and outstanding research questions
It is beyond question that the widespread prescribing of statins has resulted in significant reductions in the morbidity and mortality of cardiovascular disease. Moreover, the consensus opinion is that these drugs provide beneficial effects that are greater than would be expected from lipid lowering alone.
Despite the extensive use and high safety profile of statins, they have been found to exhibit unfavourable effects in some patients. Lowering of plasma cholesterol levels is primarily attributable to inhibition of HMG-CoA reductase in the liver, whereas the cholesterol-independent effects can feasibly affect every cell in the body. Essential roles for many derivatives of mevalonate in normal physiology suggest that global inhibition of this pathway (by HMG-CoA reductase) might explain some of the reported side effects of statins. As discussed in this review, clinical and experimental research has shown that many of the pleiotropic effects of statins are likely to be attributable to inhibition of small GTPase prenylation, predominantly inhibition of the Rho family GTPases. On the contrary, the reported detrimental effects of statins appear to be associated with other intermediates in the mevalonate pathway (see Fig. 1). For example, statins inhibit the synthesis of coenzyme Q10 (ubiquinone), whose benzoate ring structure requires a polyisoprene side chain that is synthesised from FPP (Ref. Reference Beltowski, Wojcicka and Jamroz-Wisniewska139). Q10 is present in all cells and essentially required for both mitochondrial function and its antioxidant properties. Statin-induced Q10 inhibition has been documented in animal and human studies, and has been proposed to underlie statin-induced myopathy (Ref. Reference Marcoff and Thompson140). By inhibiting mevalonate synthesis, statins also inhibit synthesis of isopentenyl pyrophosphate (see Fig. 1), which is required for activation of selenocysteine transfer RNA (Ref. Reference Moosmann and Behl141). Inhibition of this step leads to deficiency or dysfunction of selenoproteins required for muscle metabolism and hence has been associated with statin-induced muscle myopathies.
Given that the unwanted side effects of statins arise from inhibition of the mevalonate pathway, and that the beneficial pleiotropic effects of statins are perceived to be due predominantly to inhibition of Rho-family small GTPases, it is logical to investigate whether inhibition of Rho signalling itself offers clinical benefits to myocardial remodelling without the side effects of statins. Despite growing interest in ROCK as a therapeutic target, there are surprisingly few clinical trials that have used ROCK inhibitors (Refs Reference Olson142, Reference Dong143). Currently, the isoquinoline derivative fasudil (HA-1077) is the only ROCK inhibitor available for clinical use, although this agent has limited specificity for ROCK over other kinases, especially PKA (Ref. Reference Bain144). Human trials have assessed the efficacy of fasudil in stroke, angina, vascular resistance, hypertension, atherosclerosis and aortic stiffness (Ref. Reference Olson142). In addition, fasudil is currently undergoing clinical trials for treating atherosclerosis and hypercholesterolaemia (ClinicalTrials.gov identifier: NCT00120718). The potential of orally active ROCK inhibitors for treating MI, LV hypertrophy and heart failure has not yet been examined in humans (Ref. Reference Dong143). The established safety record of fasudil suggests that ROCK inhibition per se is a promising target for therapies aimed at reducing cardiac dysfunction. Other more selective inhibitors of ROCK (e.g. Y27632, GSK269962A, SB772077B, SR-715, SR-899 and SLx-2119) have not yet been assessed in man (Ref. Reference Dong143). Therefore, although new concepts are currently being explored to mimic some of the pleiotropic effects of statins, the major rationale for prescribing these drugs is that of cholesterol lowering. It remains to be proven whether there are any additional clinical advantages in developing new therapies to specifically target small GTPases.
In this review we have discussed the clinical and experimental benefits of statins on the heart and its key constituent cells, cardiomyocytes and CFs. The fact that statins exhibit properties that are distinct and nonoverlapping with current medical therapies for heart failure leads to the proposition of added benefit for patients. Understanding the mechanisms of the pleiotropic effects of statins on the heart remains an important goal if we are to expose targets for more focused therapies to reduce adverse myocardial remodelling in man. Although some such trials are in progress, there are many more avenues to be explored in the future.
Acknowledgements and funding
The authors have received British Heart Foundation funding for research on the pleiotropic effects of statins. They would like to thank the peer reviewers for positive and constructive feedback.




