


It is now about 50 years since the first recognition of a microscopic human copy number variation (CNV) – trisomy 21 (Ref. Reference Lejeune, Gautier and Turpin1) – and five years since the first reports of the widespread prevalence of submicroscopic CNVs (Refs Reference Iafrate2, Reference Sebat3) (Table 1). Classical genetics was based on the premise that all genes come in pairs, but, in the interval between these two milestones, evidence gradually accumulated to discount this dogma. The earliest examples – trisomy 21, monosomy X (Ref. Reference Ford4), and XXY (Ref. Reference Jacobs and Strong5) – had clear clinical consequences (Down, Turner and Kleinfelter syndromes, respectively), but the remarkable revelation associated with the submicroscopic CNVs has been their ubiquity throughout and among all genomes, not just those that come to medical attention. The past five years have yielded rapid developments in technology and analysis, creating a field of investigation that is transforming both our concept of the human genome and the application to clinical practice. CNVs are integral to the full spectrum of human variation and its relationship to health and disease.
Table 1. History and milestones in human copy number variation research

Definition and scope of CNV: five years later
After the seminal reports of 2004 (Refs Reference Iafrate2, Reference Sebat3), the abbreviation CNV was first formalised by Feuk et al. (Ref. Reference Feuk, Carson and Scherer6), who defined it operationally as ‘a segment of DNA that is 1 kb or larger and is present at a variable copy number in comparison with a reference genome’ (Box 1). The umbrella classification group of genomic structural variation includes CNVs as well as segments that involve no loss or gain of material but are rearranged relative to a reference (i.e. inversions or balanced translocations). Although all are biologically important and can impact phenotypes, we limit the focus of this review to matters of CNVs. Discussion has persisted as to use of ‘variant’ in this context. Notwithstanding precedents from cytogenetics and single-nucleotide terminology, our increasing awareness of the inconsistent associations between CNVs and phenotypes reinforces recommendations (Refs Reference Scherer7, Reference Lee, Iafrate and Brothman8) concerning nomenclature: to use ‘variant’ in a generic sense without inherent implications as to pathogenicity, frequency or other characteristics. It seems pragmatic to retain a term without excess denotation, and not attempt a priori to suggest that it is anything more than an observation of difference. Issues of pathogenicity or polymorphism can be addressed with modifiers (easily adapted as information arises).
Box 1. Terminology
Structural variation/variant
This is the umbrella term to encompass a group of microscopic or submicroscopic genomic alterations involving segments of DNA. We use the term as a neutral descriptor with nothing implied about frequency, association with disease or phenotype, or lack thereof. The structural variation may be quantitative (copy number variants comprising deletions, insertions and duplications) and/or positional (translocations) or orientational (inversions).
Copy number variation/variant (CNV)
CNV refers to DNA segments for which copy number differences have been observed in the comparison of two or more genomes. Without further annotation, CNV carries no implication of relative frequency or phenotypic effect. These quantitative structural variants can be genomic copy number gains (insertions or duplications) or losses (deletions or null genotypes) relative to a designated reference genome sequence.
Insertion/deletion (indel)
Indel is a collective abbreviation to describe relative gain or loss of a segment of one or more nucleotides in a genomic sequence. It allows the designation of a difference between genomes in situations where the direction of sequence change cannot be inferred: for example, when a reference or ancestral sequence has not been defined. It has typically been used to denote relatively small-scale variants (particularly those <1 kb); however, we do not propose any size restriction for its use.
Segmental duplication
This is a segment of DNA >1 kb in size that occurs in two or more copies per haploid genome, with the different copies sharing >90% sequence identity. These segments can also be CNVs. The duplicated blocks predispose to nonallelic homologous recombination.
Human genome reference assembly
The standard reference DNA sequence (or assembly) of the human genome. The assembly is derived mostly (>60%) of DNA from a single donor, with the rest of the sequence originating from a mosaic of other sources. The current assembly covers most of the euchromatic regions of the human genome.
Single-nucleotide polymorphism (SNP)
A variation in DNA that involves replacement of one nucleotide base for another is called a SNP. Polymorphism implies that the variant (minor) allele has a frequency of at least 1%; however, terminology has come to be applied more loosely by some, to include even rare mutations.
Syndrome
Literally ‘running together’, syndrome describes a collection of features or symptoms (typically comprising three or more clinical findings), the constellation of which is recognisable as a specified disorder.
Relative risk and odds ratio
Relative risk (RR) and odds ratio (OR) are similar in that they both determine the likelihood that a member of one group (individuals with a CNV) will develop a phenotype, relative to the likelihood that a member of another group (individuals without a CNV) will develop that same phenotype. For RR, this likelihood is measured using probability; for OR, it is measured using odds. With such metrics, researchers have discovered CNVs within clinical cohorts that are risk factors for distinct phenotypes.
However, we suggest that the size component of the CNV definition be reconsidered, and perhaps simply dropped. The initial limitation to segments of at least 1 kb was perhaps more a reflection of the technologies first used to reveal this class of variation [especially array comparative genome hybridisation (aCGH) with bacterial artificial chromosome (BAC) probes] than of a biological or functional threshold. Clearly, most quantitative variation in the genome involves segments smaller than 1 kb (Refs Reference Conrad9, Reference Khaja10, Reference Beckmann, Estivill and Antonarakis11, Reference Wain, Armour and Tobin12, Reference Conrad13). Thus, although ‘indels’ and di- and tri-nucleotide repeats are not technically CNVs according to the original definition, this term is now often used as a ‘catch-all’ that encompasses all non-SNP (single-nucleotide polymorphism) unbalanced variation in the genome. Thus, for some of the same reasons discussed above, we suggest that CNV be used in a less restrictive sense and be classified when needed (e.g. CNVs greater than 1 kb).
The microscopically visible CNVs at the larger end of the spectrum (1 Mb or larger) are almost invariably associated with phenotypic consequences that are likely to bring an individual to medical attention. As we move down the size spectrum, some genomic variation is of striking clinical effect, but much contributes to what we understand as normal phenotypic variation: that which simply makes individual humans different from one another (Refs Reference Buchanan and Scherer14, Reference Varki, Geschwind and Eichler15). Some CNVs are also likely to be entirely inconsequential (Refs Reference Conrad13, Reference Redon16). As with variation at the level of the individual nucleotide, many of these CNVs provide the species with a reservoir of adaptive potential for changing environmental circumstances. CNVs, therefore, are involved in every aspect of the phenotype, and whether or not a given CNV is of clinical consequence may be a function of time, place and other factors. While acknowledging this breadth of influence of CNVs, we limit the discussion for this review to examples that are likely to be relevant to medical practice now and in the near future.
CNVs have become the genomic bridge to meld disciplines of molecular genetics and cytogenetics (Table 1). The light microscope revealed the first gains and losses, starting with whole-chromosome aneuploidies, and then partial chromosome changes large enough to be obvious with solid staining. By the mid 1970s, the more indirect tools of molecular genetics, such as Southern blot hybridisation, began to expose quantitative DNA changes from the small end of the spectrum. Later, the hybridisation of molecular probes to human chromosomes, particularly with fluorescence in situ hybridisation (FISH), provided a potent tool for detection of subtle segmental deletions, duplications and rearrangements. With this and, in tandem, the DNA sequencing efforts of the Human Genome Project, segmental changes started to be recognised as a basis for many mendelian disorders as well as contiguous gene syndromes (Ref. Reference Emanuel and Shaikh17). The emergence of array technologies, particularly aCGH, facilitated widespread efficient scanning of the genome for quantitative changes in a size range that had not previously been accessible. In 2003–2004, a few studies started to observe complex CNV and structural variations at multiple loci (Refs Reference Scherer18, Reference Fredman19, Reference Cheung20); however, the Human Genome Project's strong message of 99.9% human sequence identity between two unrelated healthy individuals, with most variation encompassed by SNPs, nonetheless prevailed.
By 2004, it was apparent that CNVs are not just a cause of disease, but are ubiquitous among human genomes and an important aspect of human variation (Refs Reference Iafrate2, Reference Sebat3). Despite the profound logistical challenges associated with studying these complex genomic features, progress has been swift. As whole-genome sequences are becoming available for comparison, we foresee greater opportunity for fruitful analyses and applications in personalised medical care.
Forms of CNVs
The gain or loss of genomic material is recognised by comparison of reference and sample genomes through hybridisation or sequence analysis, and is described in relation to the reference (Fig. 1). Simple CNVs take the form of deletions, or tandem or insertional duplications. Sites at which a greater degree of replication has evolved allow a greater variety of copy number alleles among haploid genomes, with the potential for incremental variation in the related individual phenotypes. Many CNVs, however, show highly complex rearrangement of a genomic region, reflecting a history of steps in their generation, sometimes with both gain and loss of material. CNVs may involve whole genes, portions of genes, multiples of contiguous genes, regulatory elements, or none of the above, and the nature and extent of material that is deleted or duplicated is undoubtedly important for the phenotypic consequences.
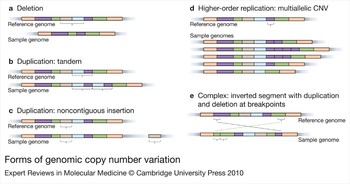
Figure 1. Forms of genomic copy number variation. Variations in sample genomes are depicted relative to a reference genome. Colours represent different segments of DNA, such that segments of the same colour contain identical sequences. Schematics show (a) deletion, or loss, of sequence (brown and blue segments) as well as (b, c) duplications of DNA segments. Duplications can be either (b) tandem, where segments (blue and purple) are duplicated into the adjacent sequence, or (c) noncontiguous, where segments (brown) can be duplicated distantly from the original sequence, even on another chromosome. The figure also shows schematics of more complicated variation, including (d) higher-order replication, where a segment (purple) can be duplicated several times and exist in multiple alleles, and (e) a complex rearrangement including an inversion (change in orientation) of sequence associated with duplication (part of the green segment) and deletion (part of the purple segment).
The nearby genomic sequence may yield clues as to how the CNV was generated (Ref. Reference Kim21). Often, a CNV is flanked by nearly identical blocks of sequence, called segmental duplications or low-copy repeats, or by Alu or LINE repetitive elements, which have created the opportunity for misalignment of DNA strands during recombination. This process of nonallelic homologous recombination (NAHR) (Ref. Reference Stankiewicz and Lupski22) was first suggested as the basis for duplications causing Charcot–Marie–Tooth disease type 1A (CMT1A) (Ref. Reference Lupski23) and subsequently for recurrent changes associated with a wide array of other genomic disorders (Ref. Reference Emanuel and Saitta24).
As more and more genomes are assayed, evidence is accumulating for other mechanisms that generate gains and losses. These include nonhomologous end joining (NHEJ) (Refs Reference Moore and Haber25, Reference Conrad and Hurles26), a process that is important for generation of B cell and T cell receptor diversity; fork stalling and template switching (FoSTeS) (Ref. Reference Zhang27), first invoked to explain nonrecurrent rearrangements in Pelizaeus–Merzbacher disease (Ref. Reference Lee, Carvalho and Lupski28) and more recently for duplications and triplications of the MECP2 (methyl-CpG-binding protein 2) gene associated with developmental delay and mental retardation in males (Ref. Reference Carvalho29); and microhomology-mediated break-induced replication (Refs Reference Hastings30, Reference Hastings, Ira and Lupski31). Although these essentially nonrecurrent mechanisms create a large diversity of CNV breakpoints with complex architecture, overlapping sets may be associated with some common phenotypic features, reflecting shared dosage-sensitive genes within the deleted or duplicated segments.
Family studies may also be informative with respect to the genesis of a CNV (as well as assessing likelihood of pathogenicity). In particular, an inversion in a parental chromosome may predispose to a de novo unbalanced variant in an offspring. The 17q21.31 microdeletion syndrome is a notable example (Refs Reference Koolen32, Reference Shaw-Smith33), as all currently reported cases result from a parental inversion common in Europeans (Ref. Reference Stefansson34). Sotos syndrome in Japanese patients is usually due to a paternal microdeletion, which is associated with a paternal inversion (Ref. Reference Visser35). Williams–Beuren syndrome is often similarly associated with predisposing parental inversions (Refs Reference Osborne36, Reference Scherer, Osborne, Lupski and Stankiewicz37) and other examples continue to emerge, reinforcing the rationale for investigation of parental samples when CNVs are found in clinical investigations, in order to properly counsel about recurrence risk.
Relationship of CNVs and SNPs
SNPs are single base substitutions found throughout the genome, each with a maximum of four possible alleles, although common SNPs usually have only two represented. They can therefore be assayed and documented in binary formats. CNVs are more complex than SNPs, often by orders of magnitude. Either form of variation can involve coding or noncoding sequences, but whereas individual SNPs affect a single site, individual CNVs may encompass multiple contiguous genes. The difference in complexity is even more important collectively, because SNPs are discrete, but CNVs among different chromosomes can be overlapping, with variable DNA portions in common and different endpoints. Furthermore, although resolution for the assays used to determine the extent (i.e. size) of CNVs has improved dramatically, there are usually still limits to the precision with which they are demarcated. As a result, it is an important but challenging task for databases to determine how to document overlapping and nested sets of CNVs in a way that is helpful for clinical research. Aside from the variable size of a CNV segment, there are aspects such as orientation and iterations to accommodate. All in all, CNVs have many more degrees of opportunity for creating variation.
The genetic relationship of CNVs and SNPs to each other (linkage disequilibrium) has been examined by determining the proportion of CNVs that can be ‘tagged’ well by nearby SNPs (Refs Reference Conrad13, Reference McCarroll38). Such ‘taggability’ was shown to depend on CNV allele frequency and local SNP density, but not CNV size. Overall, the taggability of biallelic CNVs examined was found to be largely similar to that of frequency-matched SNPs, except when rare CNVs were examined, presumably because these events were recent in origin or under negative selection. Interestingly, deletions are found to be better tagged than duplications, which may be a result of the chromosomal dispersion of some duplications and an increased frequency of reversions and multiple new mutations at some duplications.
Prevalence and frequency of CNVs
The remarkable insight of the past five years has been the extent to which CNVs are found as likely explanations, or at least highly suspect candidates for participation, in disease causation, and also their prevalence throughout all genomes, regardless of any association with pathology. Now that researchers are aware of this form of variation, searching for it has become very fruitful. The Database of Genomic Variants (DGV) (Refs Reference Iafrate2, Reference Zhang39) documents variation found in population control samples, with more than 29 000 CNVs recorded as of December 2009 (http://projects.tcag.ca/variation/) (Ref. Reference Iafrate2). Whether CNVs are more important or more abundant than SNPs as sources of human variation or disease is readily debated. However, it is clear that as a result of their size, CNVs collectively account for more of the variable genome than do SNPs (Refs Reference Redon16, Reference Levy40). The first two single human genome sequences (Refs Reference Levy40, Reference Wheeler41) provided an opportunity to look at the number of CNVs in individual genomes [relative to the haploid composites of the Human Genome Project (Refs Reference Lander42, 43) and Celera Genomics (Ref. Reference Venter44)]. Recently, we found that ~1.28% of nucleotide variation between the first individual human genome sequence (Ref. Reference Levy40) and the reference genome assembly was accounted for by CNV, far exceeding the 0.1% encompassed by SNPs (C. Lee and S.W. Scherer, unpublished).
The meaning of the term polymorphism in a genetic context has become muddled, and its use in describing structural variants might well be avoided in the interests of clarity. Descriptors such as ‘rare’ and ‘common’ in reference to a CNV apply to the frequency of a given variant rather than to the state of the locus. By convention, a rare variant has a frequency of less than 1% in a population, and this threshold is useful (but should always be specified). Many medical conditions that are relatively common in the population are clearly the result of heritable (and other) risk factors, but the search for common genetic variants to account for the majority of the heritability underlying these phenotypes has generally been unfruitful (Refs Reference Maher45, Reference Manolio46), first at the level of nucleotides and also with CNV analyses (Ref. Reference Conrad13). What is emerging, however, is evidence that multiple rare CNVs – de novo or inherited – may contribute to the genetic vulnerability for conditions such as schizophrenia or autism (Ref. Reference Cook and Scherer47), and likely to many other medically important conditions. This creates situations of great complexity to analyse and interpret, and will continue to challenge medical researchers for years to come.
Means of detection: evolution and implications
Array CGH was the technology that disclosed the large but submicroscopic CNVs, first with array probes made from relatively large DNA segments cloned in BACs. Significant refinements have ensued, such as arrays made with smaller oligonucleotide probes (for enhanced resolution), and much greater numbers of probes on each array (for denser coverage of the genome). Arrays designed to genotype SNPs are also exploited for dosage information, by looking for stretches of these markers with increased or decreased signal intensity. Recent strategic modifications to SNP arrays enhance the opportunity to discover CNVs along with concomitant SNP genotypes. The scope of these arrays may be genome-wide (with breadth but with gaps in coverage), targeted (for example to a specific gene or region of interest) or semitargeted (such as only probes for chromosome 21). Particularly for clinical diagnostics, hybrid panels are being developed, with some depth of genome-wide coverage in addition to higher density of regions known to harbour clinically relevant CNVs (Ref. Reference Carter48).
The alternative to hybridisation methods for detection of CNVs is direct comparison of DNA sequence data between reference and other genomes. As methods for whole-genome sequencing become more efficient and effective, individual genome data will soon accumulate in databases and this method of analysis will undoubtedly predominate. The direct approach to sequence comparison (Refs Reference Khaja10, Reference Levy40, Reference Wheeler41, Reference Wang49, Reference Bentley50, Reference Ahn51, Reference Kim52, Reference McKernan53, Reference Drmanac54) will eventually allow a much more complete and precise documentation of genomic variation (Ref. Reference Alkan55), but there can be technical obstacles that keep some genomic regions obscured (Ref. Reference Scherer7). Nevertheless, compared with array-based approaches, analysis by, for example, massively parallel sequencing can provide precise determination of breakpoints and copy number, will detect smaller alterations and copy-number-neutral rearrangements and (of particular importance for tumour analysis) can accommodate cellular admixture (Ref. Reference Chiang56).
How CNVs can cause disease
Both SNPs and CNVs provide the basis for phenotypic variability, which is essential for adaptive evolution. They may also be maladaptive in a particular environment, or more globally. In humans, this creates one end of a phenotypic spectrum recognised as disease (Ref. Reference Buchanan and Scherer14), bringing individuals and families to medical attention or seeking clinical intervention. These dysadaptive changes may directly involve genes, but not necessarily, and their pathogenicity can result from quantitative (dosage) or disruptive effects (Table 2; Fig. 2). CNVs that are intragenic or involve a single gene may have functional consequences that are similar to point mutations, behaving much as classical mendelian dominant or recessive traits. Alternatively, CNVs overlapping genes can result in fusion genes that may have phenotypic consequences. More extensive CNVs comprise multiple genes and underlie the ‘contiguous gene syndromes’ or genomic disorders (Ref. Reference Lupski57). Many other conditions seem to be related to complex combinations of events at noncontiguous loci.

Figure 2. Ways by which copy number variation can cause disease. This figure illustrates mechanisms underlying quantitative (dosage) or disruptive effects of copy number variation (CNV). Genes are indicated by coloured boxes, while promoters are depicted by coloured ovals. The direction of transcription is indicated by bent arrows above the genes. (a) CNVs can change the number of functional gene copies, through whole or partial deletions or duplications of genes. (b) A recessive mutant allele (indicated by red marker) can be unmasked by a deletion, which causes the loss of both functional copies of the gene. (c) Contiguous gene deletions can also eliminate (green) or disrupt (blue and red) functional genes; additionally, the mechanisms causing contiguous gene deletions can also cause a reciprocal duplication. These duplications can disrupt a dosage-sensitive gene (blue) or increase the copy number of a dosage-sensitive gene (green), which can cause disease. (In this example, another gene, shown in red, has partial duplications of its 3′ end.) (d) CNVs can also cause disease when deletions or duplications interrupt control regions that regulate juxtaposed and distant genes. Lastly, (e) CNVs can have an incremental effect when the copy number of dosage-sensitive genes is modified.
Table 2. Spectrum of copy number variation genotypes and illustrative phenotypes

aColoboma, heart anomaly, choanal atresia, retardation, genital and ear anomalies.
bWilms tumour, aniridia, genitourinary anomalies, mental retardation.
Abbreviations: CNV, copy number variation; HIV/AIDS, human immunodeficiency virus infection and/or acquired immune deficiency syndrome. Full versions of gene names can be found on the HUGO Gene Nomenclature Committee website (http://www.genenames.org/).
Deletion of a genomic segment causes hemizygosity for the deleted interval, which may also result in haploinsufficiency for dosage-sensitive gene(s). For example, a CNV deletion of LCE3B and LCE3C (two late cornified envelope genes) has been shown to be a risk factor for psoriasis (Refs Reference Hüffmeier58, Reference de Cid59). Copy number gains, such as duplications, may create imbalances due to excess product of the duplicated genes, or, when intragenic, may alter the structure of a product and thereby its function. The dosage effects of CNVs can be incremental, particularly when associated with higher-order replication (e.g. due to unequal crossover events), in which case, the relationship between copy number and disease states may be more subtle and related to thresholds (Fig. 2c and e). Other studies are revealing certain phenotypes to be associated with a more generalised increase in CNVs. For example, the number of CNVs per genome is strikingly increased in cancer-prone individuals in families with Li–Fraumeni syndrome (Ref. Reference Shlien60), and this observation has prompted similar investigations for neuroblastoma (Ref. Reference Diskin61) and many other phenotypes. CNVs are manifesting the extent to which genomes are unstable, and family studies will allow determination of not just how commonly CNVs exist, but also how frequently they occur de novo or change during transmission between generations (Ref. Reference Lupski62) (Fig. 3).

Figure 3. Complexities of de novo and inherited copy number variation. This figure uses a schematic of chromosomes (blue, paternal; pink, maternal) to illustrate transmission of copy number variation (CNV) to offspring. The gene copy number is given below each chromosome pair. Both de novo (indicated by curved arrow) and transmitted changes in CNV copy number are shown. In (a), single de novo deletion and duplication are shown within the maternal chromosome. In (b), no de novo changes are seen, but in each case the offspring has a different copy number than the parents. In the case of the multiallelic variant shown on the right, offspring have the same gene copy number but different gene configurations. Finally, in (c), both de novo and transmitted changes in copy number are combined to show a complex multilocus CNV. In this example, the offspring shows no change in copy number, despite de novo deletion.
Disruptive effects of CNVs result from a variety of mechanisms. A breakpoint within a gene may functionally disable it, but there might also be impact due to disruption or disassociation of promoters or other regulatory elements, or effects on local chromatin structure (Refs Reference Cahan63, Reference Henrichsen, Chaignat and Reymond64) (Fig. 2d). These effects may be long-range; for example, microduplication of a conserved noncoding sequence about 110 kb downstream of the BMP2 (bone morphogenic protein 2) gene, with demonstrated enhancer function, was recently shown to underlie brachydactyly type 2A in two families (Ref. Reference Dathe65). A study of gene expression in HapMap lymphoblasts revealed more than half of the effects of currently known CNV are caused not by altering gene dosage, but by gene disruption or by affecting regulatory or other functional regions, some more than 2 Mb apart (Ref. Reference Stranger66). Analysis of gene expression from within and flanking the region deleted in Williams–Beuren syndrome (Ref. Reference Merla67) found evidence of significant dysregulation of genes up to 6.5 Mb beyond the deleted region, as well as a lack of direct correlation with copy number for expression of the deleted genes. Clearly the cis-regulatory effects of CNVs can spread well beyond their borders, and genes involved in disease phenotypes may well lie outside of the associated deleted or duplicated segments.
Pathogenicity of a given CNV can be difficult to establish. In investigations initiated by an abnormal phenotype in an individual or cohort (phenotype first), the goal is to find a genotypic explanation to enhance further studies (for clinical research) or to make a diagnosis (in clinical practice) (Fig. 4). The implications of determining pathogenic potential become greater when a CNV is found before a phenotype is known (genotype first), and predictions are expected, upon which interventions may be taken – prenatal diagnosis being, of course, the circumstance of greatest concern in this respect. Various characteristics of pathogenic versus benign variants are outlined in Table 1 of Ref. Reference Lee, Iafrate and Brothman8; major considerations involve validation to confirm the chromosomal location and extent of the variation, family studies to determine whether others share the variant genotype or it is de novo, comparison with precedents documented in databases of healthy or affected individuals [such as DGV or DECIPHER (Ref. Reference Firth68), respectively] and knowledge of the genic content of the variant segment. Thus, a CNV that is inherited from a healthy parent or found in healthy family members, or that overlaps variants established in the DGV or does not involve genes of known clinical significance, is more likely to be phenotypically benign. A CNV that is shared by affected family members, or is documented in association with clinical phenotypes [found to be a risk factor using relative risk (RR) and/or odds ratio (OR) (Box 1)], or is gene-rich, particularly if any genes involved are documented in the morbid map of Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/omim/), is more likely to be of pathogenic consequence. It is important to note that the characteristics described here rarely allow a definitive determination of whether a given CNV is or is not the explanation for a phenotype (for phenotype-first investigations) or will cause a particular phenotype (for genotype-first studies). Importantly, additional functional studies would need to be conducted to investigate pathogenicity of CNVs and understand the relationship between the genotype and the phenotype. As with most areas of medicine, accurate annotation of the cause and effect of genomic variation requires a combination of analyses, experience and expert judgement for interpretation.
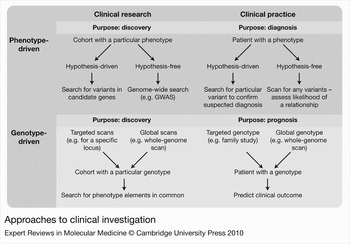
Figure 4. Approaches to clinical investigation. This figure breaks the different approaches for clinical investigations into phenotype-driven and genotype-driven approaches. These are further broken into investigations involved in clinical research, aimed at discovery, and investigations involved in clinical practice, aimed at diagnosis or prognosis. Flow charts illustrating different investigations to discover and analyse copy number variation are included in each category. The means of CNV ascertainment, be it phenotype-driven or genotype-driven, can significantly influence the interpretation of disease associations. Abbreviations: CNV, copy number variation; GWAS, genome-wide association study.
Aside from its content, the overall genomic context of a particular CNV is critical to its phenotypic consequences, and this knowledge is still in its infancy. In a very simple example, a CNV that deletes a dosage-insensitive gene may be completely recessive, but if the remaining allele happens to carry a functional mutation, then a phenotype may ensue (Fig. 2b). Research may eventually identify specific pathogenic combinations of CNVs that otherwise might be individually benign (Ref. Reference Lee, Iafrate and Brothman8), or the converse: certain CNVs that are pathogenic unless a compensatory element is present elsewhere in the genome, epigenome or environment to reduce penetrance. Interpretations of this kind will be enhanced as whole-genome sequence analysis becomes the norm. The apparent lack of phenotype associated with an isolated CNV does not rule out its pathogenic potential in another genomic context. Although a CNV that has arisen de novo is more likely to be pathogenic than one that has escaped selection in a family or population, this is again probabilistic. This assertion is due to the fact that inherited CNVs are present in at least one reproductively viable individual, while de novo CNVs are present in a single individual and may not have been subjected to negative selection. However, this is not a definitive distinction between these classes of CNVs. Finally, CNVs of pathological consequence are more likely to be large (encompassing many genes and/or regulatory sequences), and to involve loss, rather than gain, of genomic material (although early data are somewhat biased because of the relative ease of ascertainment of deletions and larger segments) (Ref. Reference Conrad13).
Means of ascertainment shapes findings
Ascertainment bias is an inevitable component of research, and acceptable as long as it is acknowledged and accounted for. The remarkable aspect of CNVs has been not so much discovery of their association with genomic disorders as their ubiquity throughout genomes of general populations. To study cause-and-effect relationships and to put knowledge to use for clinical practice, we need to compare the prevalence of CNVs between defined cohorts and nonclinical samples. The developing means to undertake relatively hypothesis-free and fully comprehensive data collection has begun to provide unprecedented opportunities for such analysis (Fig. 4). For a fully genotype-first approach, data must be gathered from an unselected cohort, such as all newborns, followed by phenotypic comparison of all those who share particular CNV genotypes. A phenotype-first approach involves collecting a cohort with a particular clinical presentation or diagnosis, and looking for CNVs that are more prevalent among them, relative to those without the phenotype. Genotyping can be genome-wide (hypothesis-free), or a targeted search around a candidate locus [compare, for example approaches of Miller et al. (Ref. Reference Miller69) and Sharp et al. (Ref. Reference Sharp70) with respect to the 15q13.3 deletion syndrome]. Many current studies are somewhat intermediate, in that they involve samples referred for analysis because of some clinical finding, and research outcomes can be strongly influenced by reasons for such referrals – such as developmental delay, behaviour issues, dysmorphic features and so on. A recent commentary (Ref. Reference O'Donovan, Kirov and Owen71) compares early phenotype-led and more-recent CNV-led studies (Refs Reference Brunetti-Pierri72, Reference Mefford73) that focus on phenotypes associated with deletions and duplications at 1q21.1, with markedly different outcomes from the various approaches – what you find depends largely on what you look for.
Penetrance and expressivity of CNVs
A mutant gene is described as fully penetrant when all individuals with the mutation express the related phenotype, whereas reduced penetrance refers to a situation in which some individuals with a given genotype show phenotypic evidence of it and some do not. To some extent, it is one end of a spectrum of variable expression of the phenotype, and the concepts are inter-related. They apply also to observations about the much more complex genotypes involving CNVs and their associated phenotypes, and examples span a wide range of relationships. In some situations, there is a consistent relationship between the CNV genotype and at least a core phenotype, such that all individuals with (for example) a given deletion share a definable phenotype, and every individual with that phenotype has a similar or overlapping deletion. Clinical examples of this kind include Williams–Beuren syndrome, Prader–Willi and Angelman syndromes, and the 17q21.31 deletion syndrome. Other CNVs, such as the most common human microdeletion – 22q11.21 – are highly penetrant but with a range of phenotypic expression so broad as to encompass more than one clinically designated syndrome (Refs Reference Carlson74, Reference Driscoll75). A nearby multiply ascertained microduplication was associated with such disparate findings as to ‘obfuscate the clinical relevance of the molecular data’ (Ref. Reference Coppinger76), and the 1q21.1 microdeletions are said to be so variable as to ‘elude syndromic classification’ (Ref. Reference Mefford73). Other conditions, notably psychiatric disorders including autism, have a more nuanced connection to the various CNVs emerging as factors that are significantly associated but not independently causative for the phenotype (Ref. Reference Cook and Scherer47). Evidence of reduced penetrance abounds in these families with inherited CNVs, although retrospective evaluation of apparently unaffected parents or other relatives sometimes reveals subtle features of the proband's phenotype.
Buffering or modifier effects have been described for the thrombocytopaenia absent radius (TAR) syndrome (Refs Reference Klopocki77, Reference Uhrig78), for which a deletion at 1q21.1 is necessary but not sufficient to cause the syndrome, and for spinal muscular atrophy (Ref. Reference Prior79), where the impact of an intragenic deletion of the SMN1 (survival of motor neuron 1, telomeric) gene may be tempered by normal variation in the number of gene copies of SMN1 and the closely related SMN2. Epigenetic effects may also influence expression of the phenotype, as exemplified with Silver–Russell syndrome (Ref. Reference Schonherr80) and possibly developmental verbal dyspraxia (Ref. Reference Feuk81).
Germline and somatic CNVs
Genomic alterations, including CNVs, have one of four origins: they can be (1) inherited from a parent with the same germline variant, (2) inherited from a parent with germline mosaicism for the same variant, (3) arise de novo from a parental germ cell or (4) arise de novo in a somatic cell (Fig. 3). The latter category is especially relevant to the field of cancer genomics because of generalised genomic instability, and clonal expansion and evolution of tumour cells. Somatic mosaicism for CNVs has also been noted in monozygotic twins (Ref. Reference Bruder82) and in different tissues of an individual (Ref. Reference Piotrowski83), as well as in diseases such as Rubinstein–Taybi syndrome (Refs Reference Gervasini84, Reference Roelfsema and Peters85, Reference Schorry86), tuberous sclerosis (Ref. Reference Kozlowski87) and aniridia (Ref. Reference Robinson88). In clinical genetics applications, it is important to distinguish among these categories, both for clinical research and to predict outcomes for newly ascertained probands and recurrence risks for families. Among de novo events, some are truly random, but, in contrast to single-base mutations, the structural variants are often associated with vulnerable genomic regions in which similar CNVs tend to recur. These variants can, in turn, beget more genomic instability with disrupted chromatin structure or opportunities for misalignment.
Enigmas in CNV genotype–phenotype relationships
Some approximately similar CNVs have emerged in the context of various different complex phenotypes. Duplications of 17p12 cause CMT1A and the reciprocal deletion is associated with hereditary neuropathy with liability to pressure palsies (HNPP), but the region is also implicated in schizophrenia (Ref. Reference Kirov89). Deletions at 1q21.1 also emerged in schizophrenia phenotype-driven studies (Ref. Reference Kirov89), but CNVs of this region are found enriched in association with phenotypic features such as micro- or macrocephaly, mental retardation, cardiac anomalies or autism (Refs Reference Brunetti-Pierri72, Reference Mefford73). Furthermore, family studies demonstrate that the same CNVs can be without apparent consequence in some individuals.
Some ostensibly similar phenotypes are associated with various different genotypic findings, each of a magnitude to elicit suspicion with respect to pathogenicity. Neurological and psychiatric conditions seem to predominate as examples (Table 2) but the cardiac defect known as tetralogy of Fallot has recently provided similar genotypic characteristics (Ref. Reference Greenway90).
Syndrome, meaning ‘running together’, describes clinical entities that involve constellations of features from different systems. Certainly there are examples of single-nucleotide mutations that have pleiotropic effects and create multisystem phenotypes, but CNVs are more likely to do so because of their potential to compromise multiple genes, with concomitantly widespread effect. As more information emerges about such genotype–phenotype relationships, we are struck by the enigma that some classical syndromes, Williams–Beuren syndrome for example, have a relatively consistent genotype and phenotype presentation, whereas the highly recognisable phenotypic constellation of Down syndrome can result from CNVs ranging from full trisomy 21 to almost any portion thereof. At the same time, some recurrent CNVs have been discovered in clinically unselected cohorts (such as microdeletions of 1q21.1, described above) that, despite considerable genotypic consistency, have no recognisable consistent ‘running together’ of features.
Clinical exemplars
The ability to undertake whole-genome scans by arrays or sequencing has provided the opportunity to discover individual disease-associated CNVs in the absence of any prior hypotheses as to their chromosomal location. This holistic approach is also revealing combinations of CNVs, both collectively and within individuals, that may become the key to understanding complex phenotypes such as autism, bipolar disorder, schizophrenia, macular degeneration, or tetralogy of Fallot (Table 2). A concept is emerging of CNV load, as cohorts or individuals are recognised to have a higher than average number of CNVs, rather than specific aberrations in candidate genes. Li–Fraumeni syndrome provides a striking prototype (Refs Reference Shlien60, Reference Need91). Below, we describe representative examples of the effects of CNVs in clinical conditions.
Down syndrome
Down syndrome is something of a metaphor for the progress of CNV discovery in humans. When we consider CNVs in the broader definition to include microscopic variants, then trisomy 21 was arguably the first to be discovered (Ref. Reference Lejeune, Gautier and Turpin1) (Table 1). Whole-chromosome aneuploidies have different underlying mechanisms than the submicroscopic variants, but the phenotypic consequences are not categorically distinct; rather, they are part of a continuous spectrum in this respect. After recognition of nondisjunctional trisomy 21, rearrangements such as Robertsonian translocations were found as the basis for duplicated long arms of chromosome 21, and then microscopic partial trisomies, followed by those detectable by FISH. Eventually, arrays have been used to fine-tune the extent of duplicated material with higher-resolution mapping, and to study correlates of specific features of the phenotype with particular genes or regions (Refs Reference Lyle92, Reference Korbel93). A very early study of the reciprocal deletion syndrome (i.e. partial monosomy 21) made some prescient observations concerning gene-dosage effects: ‘Our findings do add weight to the hypothesis that genetic control of enzymes is not a simple gene-dosage affair, but a complex interaction of structural, regulator, and modifying genes which may be located at various loci on different chromosome segments.’ (Ref. Reference Reisman94).
Williams–Beuren syndrome and its reciprocal 7q11.23 duplication syndrome
Williams–Beuren syndrome is one of the classic genomic disorders – a contiguous gene syndrome associated with a recurrent microdeletion of 7q11 that is strikingly consistent. The recurrence is mediated by flanking segmental duplications and by a relatively common inversion of the region (carried by up to a third of parents of affected individuals and 5% of the general population), which predisposes to aberrant meiotic recombination in parental chromosomes, with pathological outcomes in the offspring (Refs Reference Osborne36, Reference Ewart95, Reference Osborne and Mervis96). The deletion phenotype is a relatively predictable syndrome. As anticipated for the CNVs mediated by NAHR, by which deletion outcomes should be matched by reciprocal duplication products (Refs Reference Stankiewicz and Lupski22, Reference Lupski97), the complementary duplication syndrome was eventually recognised (Refs Reference Somerville98, Reference Kriek99, Reference Torniero100). Its clinical phenotype is distinct from that associated with the deletion, and, particularly with respect to expressive speech ability, is in striking contrast, suggesting some effects of gene dosage. As discussed, the impact of the Williams–Beuren microdeletion extends to genes well beyond the borders of the aberrant segment (Ref. Reference Merla67).
15q13.3 microdeletion and duplication phenotypes
This recurrent CNV locus has been recognised only recently, by aCGH (Refs Reference Sharp70, Reference Sharp101), but is repeatedly coming to attention from a variety of study groups. It illustrates the challenges in assessing pathogenicity of these variants, and the impact of ascertainment. The region is adjacent to that deleted in Prader–Willi and Angelman syndromes (PWS/AS), which together feature a series of duplication blocks demarcated by recurrent breakpoints (BP1 to BP6) (Refs Reference Christian102, Reference Mignon-Ravix103, Reference Sahoo104). Just distal to the PWS/AS region is the 1.5 Mb segment BP4–BP5, which is found to be deleted or duplicated in an increasing number of individuals ascertained through routine and targeted clinical investigations, occasionally as part of a larger CNV (Refs Reference Sharp105, Reference Stefansson106, Reference Pagnamenta107, Reference Shinawi108, Reference Helbig109). Clearly the region is enticing, drawing interest from several clinical directions, but observations are disparate. Among controls, deletions have been mostly limited to a handful of Icelandic individuals (Ref. Reference Stefansson106), but several studies that included family investigations have discovered deletions or duplications in parents or other relatives who do not share the probands' phenotypes (Refs Reference Miller69, Reference Ben-Shachar110, Reference Pagnamenta111, Reference van Bon112), indicating that deletion of this region is not necessarily pathogenic, but also not inconsequential.
Even from relatively untargeted clinical investigations (Refs Reference Miller69, Reference Sharp70, Reference Ben-Shachar110, Reference van Bon112), details of phenotypes associated with the 15q13.3 CNVs are skewed by the nature of the referral base – for example, predominantly developmental delay, dysmorphic features, multiple congenital anomalies and behaviour issues. Deletions of 15q13.3 were found in up to 0.3% of such referrals; duplications were rarer. When parental samples were available, the majority of these probands' CNVs were found to be inherited.
In studies of more clinically defined cohorts (phenotype-first), deletions of the BP4–BP5 segment (or more) were rarely, but significantly, associated with schizophrenia (Refs Reference Stefansson106, Reference Consortium113) or idiopathic generalised epilepsy (Ref. Reference Helbig114); deletions or duplications of the same segment appear with tantalising frequency when autism or related features such as expressive language delay are part of the phenotype (Refs Reference Miller69, Reference Ben-Shachar110, Reference Pagnamenta111, Reference van Bon112). Despite relative consistency of the CNV genotypes found across a broad range of studies, the phenotypes of these individuals vary greatly. With current evidence, the finding of a CNV involving 15q13.3 in a clinical investigation would raise concern, but would probably be insufficient to explain or predict any particular phenotype. Such observations of uncertainty will consume a great deal of health professionals' time for the foreseeable future (Refs Reference Buchanan115, Reference Ali-Khan116).
Chromosome 1q21.1 CNV phenotypes
Evidence of a potentially contiguous gene syndrome at 1q21.1 was first noted in a targeted candidate gene study of a cohort with congenital heart defects (Refs Reference Redon16, Reference Christiansen117). Later, genome-wide phenotype-first surveys detected significant association of similar duplications with autism (Ref. Reference Szatmari118) and deletions with schizophrenia (Refs Reference Stefansson106, Reference Consortium113). Using the complementary genotype-first approach, two large studies (Refs Reference Brunetti-Pierri72, Reference Mefford73) started with relatively unselected clinical referral cohorts to ascertain, through data from genome-wide or targeted assays, large numbers of individuals with CNVs involving 1q21.1 and then to document the scope of associated clinical phenotypes. Both found large (~1.35 Mb) recurrent deletions of the region as well as reciprocal duplications, but other than some relationship between CNV dosage and micro- or macrocephaly (Ref. Reference Brunetti-Pierri72), there was such a wide range of clinical presentation among index cases that no common manifestations of a syndrome could be recognised. Furthermore, although deletions and duplications of this region are clearly rare in the general population (Ref. Reference Mefford119), family studies for those ascertained in the clinical cohort showed many CNVs to have been inherited from parents with milder or absent features relative to those of their respective offspring. Similar to the situation for 15q13.3, and undoubtedly for many regions yet to be characterised, the finding of a CNV in this region would raise legitimate suspicion, but, with current information, would be sufficient neither to explain nor to predict a particular clinical outcome.
Attention has been drawn recently to a smaller previously known CNV (Ref. Reference Pinto120) just distal to the 1q21.1 region, for which the deletion allele is common (9.1%) in controls but significantly more prevalent (15.6%) among cases with neuroblastoma (Ref. Reference Diskin61). Cis and trans effects appear to be involved as part of dosage effects on susceptibility, and scrutiny as a result of the initial observation led to discovery of a novel transcript of interest from within the deletion interval.
The 1q21.1 region is flanked proximally by another segment of 200 kb that is deleted in all individuals with TAR syndrome and not in controls studied to date (Ref. Reference Klopocki77). In at least two families (Refs Reference Klopocki77, Reference Uhrig78), the microdeletion was inherited from an unaffected parent, indicating that the CNV is necessary but not sufficient to cause the syndrome. At present, therefore, this CNV is a helpful diagnostic tool in the context of other clinical findings, but is not in itself predictive of the TAR phenotype. We also note that larger deletions of 1q21.1 could also influence risk for neuroblastoma (Ref. Reference Diskin61) and that chromosomal inversion encompassing the 1q21.1 region has also been observed (Ref. Reference Redon16).
Immunity and autoimmunity
Early observations on the impact of CNVs was that they are particularly prevalent among genes that have a role in our interface with the environment (Refs Reference Armengol, Rabionet and Estivill121, Reference Schaschl, Aitman and Vyse122, Reference Ionita-Laza123), such as those that are part of the immune system. Various gene families, such as the major histocompatibility locus, immunoglobulins, chemokines, receptors, defensins and interleukins, might reflect CNV events throughout evolution, but are also characteristically polymorphic with much of the variation contributed by CNV for individual loci. By contrast to simple deletion variants, these sites are typically multiallelic, reflecting a wide range of copy number and creating particular challenges for discerning their incremental effects and specifying exact copy numbers, but new approaches have been reported (Refs Reference Hollox, Detering and Dehnugara124, Reference Nuytten125).
Disorders with an autoimmune component, such as psoriasis (Ref. Reference de Cid59), systemic lupus erythematosus (SLE), type 1 diabetes, and rheumatoid arthritis, are all beginning to yield some of the mystery of their respective causes as more refined data emerge from these highly variable genomic sites. Interestingly, a recent discovery found that a polymorphic deletion variation upstream of IRGM (immunity-related GTPase family, M) was associated with Crohn disease (Ref. Reference McCarroll126).
Autoimmunity is a maladaptive consequence of an adaptive immune system, and variation at some of the relevant loci can have a spectrum of clinical consequences. For example, a lack of the chemokine receptor CCR5 or an excess of its ligand CCL3L1 appears to be protective against human immunodeficiency virus (HIV) infection and acquired immune deficiency syndrome (AIDS) (Refs Reference Gonzalez127, Reference Kulkarni128, Reference Shostakovich-Koretskaya129), but at the same time is associated with an enhanced inflammatory and autoimmune response, predisposing to rheumatoid arthritis (Ref. Reference McKinney130). This example reminds us that these variants do not act in isolation but as part of functional networks, and powerful analytical tools will be needed in order to interpret clinical data in the appropriate context.
Given the weak effect of HLA (human leukocyte antigen) matching to predict acute organ rejection in lung transplantation, a recent study (Ref. Reference Colobran131) considered other genetic risk factors – in particular, the chemokine ligand CCL4L1, genes for which are within a CNV region on chromosome17q12 that also contains CCL3L1. Copy number for CCL4L1 was significantly greater in patients who experienced acute rejection, and even greater in those with multiple rejection episodes than among those who did not reject their allograft. Another study (Ref. Reference McCarroll132) found a similar result when looking at mismatches for the homozygous deletion of UGT2B17 between donors and recipients, which increased the likelihood of graft-versus-host disease. Whether these observations are a direct effect of gene dosage, or a proxy for nearby variable elements is not yet clear, but undoubtedly they will spark a flurry of research activity.
Modifier effects in Li–Fraumeni syndrome
This familial cancer syndrome is caused by mutations in the TP53 gene (encoding p53), but the breadth of variation in severity, onset and types of tumours, even among those who share the same TP53 mutation, prompted a genome-wide search for evidence of modifier loci. Rather than specific genomic sites, the number and size of CNVs was found to be markedly increased in TP53 mutation carriers, particularly those with cancer (Ref. Reference Shlien60). The instability associated with these CNVs might in turn be the precursor to somatic changes and tumour formation. Information about the CNV load in TP53 mutation carriers may provide an adjunct for risk prediction and counselling (Ref. Reference Shlien and Malkin133).
A specific modifier was found within the TP53 gene, comprising a common (~10–30%) 16 bp microduplication in intron 3 (TP53PIN3), the presence of which is associated with an average onset of tumour diagnosis 19 years later than in mutation carriers without the duplicated variant (Ref. Reference Marcel134).
Two recent reports (Refs Reference Schwarzbraun135, Reference Adam136) describe deletions of 17p13.1 encompassing the TP53 gene found from whole-genome scans in three patients with mental retardation and dysmorphic features. In addition to providing a likely explanation for the referring clinical features, they predicted a Li–Fraumeni phenotype for which appropriate risk management could be recommended. These add to a larger series found from among general clinical referrals of CNVs that involve genes with probable predisposition to various cancer syndromes (Ref. Reference Adams137).
Discussion: clinical implications and applications
How are CNVs changing clinical practice?
The most conspicuous effect of the discovery of CNVs has been in laboratory medicine. There are many more diagnoses being made, but the distinction between classical cytogenetics and molecular diagnostics has become blurred (Ref. Reference Speicher and Carter138) as the gap in resolution of analysis is taken up with knowledge of this prevalent form of variation. New laboratory tools and skills are being invoked, and practitioners must broaden their expertise to encompass the entire spectrum of variation. The very particular skill of reading a traditional karyotype is rapidly being usurped by diagnostic arrays with less subjective interpretation, enhanced resolution and competitive costs. From the other end of the spectrum, awareness of interactions among single-nucleotide alterations and structural variations is increasing demand for follow-up diagnostic assays and enhancing the expectation for more comprehensive analysis. As whole-genome sequencing eventually becomes routine, the needed interpretive skills will change yet again.
New awareness of the widespread nature of this form of genomic variation reminds us of the ongoing need for healthy scepticism in diagnostic and predictive analyses – the simplest example being that apparent homozygosity for a SNP may in fact be hemizygosity, where both a SNP and a deletion are present in combination, but on different haplotypes. Only the haplotype containing the SNP would be detected by traditional genotyping assays, leading to the misclassification of the allele as homozygous. For example, in one case, a patient with cystic fibrosis (autosomal recessive) was apparently homozygous for the F508del mutation. This was unremarkable until the mother's sample tested negative for the same mutation; subsequently, she and the newborn were found to share a large deletion that encompassed the same exon (Ref. Reference Stuhrmann139). In another, more complex example, a newborn with strong clinical evidence of cystic fibrosis was negative for all standard sequence-based mutation screens, and only with quantitative assays did the laboratory find large intragenic deletions on each of the patient's CFTR alleles (Ref. Reference Girardet140). Recent evidence from global newborn screening programmes demonstrates that larger intragenic deletions of CFTR may account for 1–3% of mutant chromosomes (Ref. Reference Tomaiuolo141), or more, as awareness permeates and appropriate screening assays for CNVs are invoked (Ref. Reference McDevitt and Barton142).
For clinicians, CNVs have opened up analytical potential for clinical cases that had previously eluded diagnosis. This potential is creating a huge demand for laboratory tests that are still expensive, and very time-consuming to interpret. Nonetheless, when informative, such results may allow many patients and families the satisfaction of an explanation for their observed challenges, sometimes after years of fruitless investigations. These additional tools may allow earlier diagnosis for conditions, such as autism, for which early intervention in some individuals may be particularly beneficial.
Attention is drawn to genes of interest by virtue of their location in a newly recognised CNV [e.g. CHARGE syndrome (Ref. Reference Vissers143)], and this is opening a floodgate of research potential into complex disorders. Eventually, of course, we hope to find therapeutic prospects among such genes, and awareness of their involvement in a given phenotype is the first step. Particularly because of the tendency of larger CNVs to encompass contiguous genes, we are gaining insight into syndromology, with some improvement in explaining the spectrum of variation and the degree of consistency or inconsistency among phenotypic features. As illustrated by CNVs such as at 1q21.1 and 15q13.3 (discussed in this review), and more recently at 16p11.2 [in autism (Refs Reference Fernandez144, Reference Kumar145, Reference Marshall146, Reference Weiss147), developmental delay (Ref. Reference Shinawi148), schizophrenia (Ref. Reference McCarthy149) and obesity (Ref. Reference Bochukova150)], this opportunity can also be a Pandora's box. How CNV results are applied to research or medical decision-making needs to be weighted according to the circumstances where it is observed. For example, the relevance of the results will differ if a CNV is uncovered in (Reference Lejeune, Gautier and Turpin1) a known disease gene, (Reference Iafrate2) in a high-risk setting (such as during prenatal complications), (Reference Sebat3) through a targeted list (such as an individual with a family history of a disease or as confirmation of an existing clinical diagnosis), and (Reference Ford4) in a universal population screen (Refs Reference Buchanan and Scherer14, Reference Cook and Scherer47).
Conclusion: research in progress and outstanding research questions
Five years since the first rudimentary scans drew our attention to the widespread presence of genomic CNVs, they have become the focus for a myriad of surveys, both genotype- and phenotype-driven. Compendia such as the DGV are being refined and updated regularly. The apparent size of CNVs is decreasing as tools with enhanced resolution allow more precise definition of breakpoints, and annotation of precise copy number is becoming feasible (Ref. Reference Perry151). The complexity of these data makes them somewhat recalcitrant, and the means for documentation in an unambiguous and functional way has been significantly challenging. Even more daunting, however, is annotation of the phenotypes of individuals who do and do not carry these variant genotypes, and finding ways to merge the plethora of disparate observations.
In addition, as technologies advance, the ability to detect CNVs in an individual genome increases. This is evidenced by recent diploid genome sequencing projects that find many CNVs that are unique to an individual (Refs Reference Levy40, Reference Wheeler41, Reference Wang49, Reference Bentley50, Reference Ahn51, Reference Kim52, Reference McKernan53, Reference Drmanac54). Many of these CNVs are large and represent potentially pathogenic variation. As the cost of genome sequencing decreases, the prevalence of such studies will increase. Importantly, sequencing technologies have the potential of combining both SNP and CNV detection strategies into a single analysis, which should increase the power to detect variation that is related to phenotypes and disease.
As the HapMap project followed quickly on the heels of the first consensus genome sequence, so too has considerable effort moved to the study of how these structural variants are differentially distributed among global populations (Refs Reference Conrad13, Reference Redon16, Reference Zogopoulos152, Reference Jakobsson153, Reference Armengol154, Reference Matsuzaki155, Reference Yim156, Reference Brookes157). How does knowledge of such distributions influence the clinical interpretation of data? Can this information tell us anything about different environmental pressures to which these genomic alterations may have provided the means for adaptation? Are the phenotypic consequences different under different environmental circumstances?
Much has already been learned about mechanisms that underlie these genomic rearrangements, and the extent to which they are recurrent or randomly generated, inherited or de novo, and stable or unstable (Ref. Reference Stankiewicz and Lupski158). This has done little, however, to enlighten us about what determines whether a given CNV will have any pathogenic consequences, or will be associated with a pattern of features that might be recognised as a syndrome.
Major challenges
The connections between genomic observations and clinical implications are not straightforward, and involve complex network relationships. Single-gene disorders will continue to present themselves for medical attention, but the more prevalent and problematic conditions – heart disease, cancer, psychiatric and behavioural disorders, developmental delay, dysmorphic syndromes – require a shift in mindset from genetic-based to genomic-based. Candidate gene searches within a CNV (or group of CNVs) will need to progress to analyses of added dimensions, including gene and protein pathways and networks, for which sophisticated bioinformatics tools will be essential. Moreover, a more complete understanding of CNV and SNPs will be required to better empower genome-wide association studies (GWASs) of disease.
In our recent study, we explored whether CNVs might be plausible candidates for known complex trait associations from SNP-based GWASs. However, we found that CNVs might explain less than 5% of previously reported GWAS hits, suggesting that common CNVs are not likely to account for a large part of the ‘missing heritability’ (Ref. Reference Maher45) from complex traits. These results also emphasise the need to consider all classes of variation (CNVs, other structural variants and SNPs, both common and rare) in order to maximise power to detect causal variation in disease association studies.
As formidable as data gathering may be for these CNVs of higher-order complexity, interpretation is far more of a challenge. Examples cited in this review provide some illustration of why particular caution is needed in moving between research findings and applications in a clinical context, in particular when studying complex disease. More studies examining CNV mutation rates across chromosomes and the effects of such events on gene dosage and the functional consequences would be beneficial. Undoubtedly, with accumulation of much more information, patterns will emerge to make some sense of what can currently seem, for some CNVs and phenotypes, like an uninterpretable mass of raw data.
We will be challenged to move beyond the obvious benefit of CNVs for explaining (diagnosing) various phenotypes to their utility in prediction and prognosis. A difficulty is that the plethora of CNV data can be provided as information, but without knowledge, and healthcare providers may be burdened for some time with the ‘variant of unknown significance’.
Finally, the challenge will be not only to use our knowledge of these variants for explanation and prediction of medically relevant conditions but also to find ways to mitigate their untoward impact, for prevention or treatment of genomic disease. This will require a new level of inspired creativity.
Acknowledgements and funding
We acknowledge Dr Janet Buchanan and Dr Andrew Carson for significant contributions in preparing this review. The work is supported by Genome Canada/Ontario Genomics Institute, the Canadian Institutes of Health Research (CIHR), the McLaughlin Centre for Molecular Medicine, the Canadian Institute of Advanced Research, the Hospital for Sick Children (SickKids) Foundation and the National Institutes of Health (NIH)/National Human Genome Research Institute. S.W.S. holds the GlaxoSmithKline-CIHR Pathfinder Chair in Genetics and Genomics at the University of Toronto and Hospital for Sick Children. We also thank the peer reviewers for their helpful comments and suggestions.






