I. INTRODUCTION
Multiferroic oxides materials have combined electric, magnetic, and/or structural order parameters that result in simultaneous ferromagnetic, ferroelectric, and/or ferroelastic behaviors. The discovery of colossal magnetoresistance (CMR) in an ordered structure Sr2FeMO6 (M = Mo, W) has resulted in many recent research in double perovskite oxides with general formula A2BB'O6 (Kobayashi et al., Reference Kobayashi, Kimura, Sawada, Terakura and Tokura1998; Dass and Goodenough, Reference Dass and Goodenough2001; García-Hernández et al., Reference García-Hernández, Martínez, Martínez-Lope, Casais and Alonso2001; Ray et al., Reference Ray, Kumar, Majumdar, Sampathkumaran and Sarma2001; Branford et al., Reference Branford, Clowes, Bugoslavsky, Miyoshi, Cohen, Berenov, MacManus-Driscoll, Rager and Roy2003; Philipp et al., Reference Philipp, Majewski, Alff, Erb, Gross, Graf, Brandt, Simon, Walther, Mader, Topwal and Sarma2003; Sher and Attfield, Reference Sher and Attfield2006; Dhahri et al., Reference Dhahri, Dhahri and Oumezzine2009; Gong et al., Reference Gong, Chen and Liu2014). The ordering of Fe and Mo or W cations in the octahedral B-sites in the cubic high-symmetry structure double perovskite lattice leads to ferrimagnetism with a high Curie temperature (T C) of ≈ 420 K for Sr2FeMoO6 (Kobayashi et al., Reference Kobayashi, Kimura, Sawada, Terakura and Tokura1998), while for Sr2FeWO6 it is known to be antiferromagnetic insulator with T N ≈ 37 K (Kawanaka et al., Reference Kawanaka, Hase, Toyama and Nishihara2000), it has contrasting transport properties.
Low-field magneto-resistance emerges from spin-dependent electrons transfer to grain or magnetic domain confines. This property, related to the variation of electric resistivity when a magnetic field is applied, is a consequence of the half-metallic character of the double perovskite (Philipp et al., Reference Philipp, Majewski, Alff, Erb, Gross, Graf, Brandt, Simon, Walther, Mader, Topwal and Sarma2003). Early, the CMR materials of perovskite oxides have prospects of great opportunities for the development of new technologies, potentially applied including nonvolatile magnetic random access memory, magnetic read/write heads for hard drives, magnetic tunnel junction, and other magnetic sensor applications. Among the explored analogues of Sr2FeMO6 (M = Mo, W), it was reported that A 2CrMoO6 (A = Sr, Ba) (Arulraj et al., Reference Arulraj, Ramesha, Gopalakrishnan and Rao2000; Sher and Attfield, Reference Sher and Attfield2006) is also a ferrimagnet with a high Curie temperature of ≈ 450 K (Arulraj et al., Reference Arulraj, Ramesha, Gopalakrishnan and Rao2000). The partial disorder of Cr and Mo in the B-sites of double perovskite offers an important role in the structural, electrical and magnetic properties of these compounds (Arulraj et al., Reference Arulraj, Ramesha, Gopalakrishnan and Rao2000; Sher and Attfield, Reference Sher and Attfield2006).
As mentioned the disorder in the B-site has an important role in the behavior of physical properties. Extensive studies have been conducted to ameliorate, and/or explain this antisite-disorder and its effects on the properties, by the way of synthesis procedures or theoretical calculation (density functional theory: DFT) and the structural identification by means of Rietveld refinement. Also using magnetic measurements as a sensitive tool to demonstrate the antisite-disorder and to understand the ferromagnetic, magnetic and magneto-electric behavior (Sánchez et al., Reference Sánchez, Alonso, García-Hernández, Martínez-Lope, Martínez and Mellergård2002; Ivanov et al., Reference Ivanov, Nordblad, Eriksson, Tellgren and Rundlöf2007).
As the synthesis of Sr2AlMO6 (M = Nb, Ta) double perovskite (Woodward et al., Reference Woodward, Hoffmann and Sleight1994) which have been prepared by several different methods shows that the order degrees of octahedral site cations are obtained depending on the synthesis conditions. The series of Ni1−xCoxCr2O4 spinels were also reported as antisite-disordered compound with optical and magnetic properties (GAO et al., Reference Gao, Chang, Wu, Wang, Pang, Liu, Zhu and Yun2017). Some of the A sites are occupied by Cr+3 ions and some of the B sites are occupied by Ni and Co cations. This departure from the normal spinel structure causes the antisite defect. These antisite defects play an important role in ions surface interaction process and influence the electrical and optical behaviors. The antisite-disorder was also observed in several triple perovskites, such as Sr3Fe2TeO9 (S space group I4/m), Ba3Fe2TeO9 (space group P63/mmc) (Ivanov et al., Reference Ivanov, Nordblad, Eriksson, Tellgren and Rundlöf2007; Tang et al., Reference Tang, Hunter, Battle, Sena, Hadermann, Avdeev and Cadogan2016), Ca3Fe2WO9 (space group I4/m) (Ivanov et al., Reference Ivanov, Eriksson, Tellgren and Rundlöf2005), Sr3Fe2MoO9 (space group I4/m), Sr3Fe2WO9 (space group I4/m), Sr3Fe2UO9 (space group I4/m) (Viola et al., Reference Viola, Augsburger, Pinacca, Pedregosa, Carbonio and Mercader2003), Ba1.6Sr1.4Fe2WO9 (space group P63/mmc) (Manoun et al., Reference Manoun, Benmokhtar, Bih, Azrour, Ezzahi, Ider, Azdouz, Annersten and Lazor2011). Recently perovskite series Sr3−xCaxFe2TeO9 (0 ⩽ x ⩽ 1) (El Hachmi et al., Reference El Hachmi, Manoun, Tamraoui, Mirinioui, Abkar, El aamrani, Saadoune, Sajieddine and Lazor2017) were reported by our research team showing that the antisite-disorder is affected with increasing the Calcium amount from x = 0 to x = 1.
Using solid state reaction as synthesis technique, two perovskite compounds Ca+2 and Pb+2 doped Sr3Fe2WO9, and Sr3Fe2TeO9 respectively, are reported in this work with general formula Sr2CaFe2WO9 and Sr2PbFe2TeO9. Using X-ray powder diffraction (XRPD) the compounds are easily identified to adopt the perovskite structure. The refined crystal structure was reported and the antisite-disorder in the B and B′ sites was discussed.
II. EXPERIMENTAL
A. Synthesis
Polycrystalline powders of Sr2CaFe2WO9 and Sr2PbFe2TeO9 were synthesized using the standard solid-state reaction from stoichiometric mixtures of SrCO3 (99.995%), Ca(OH)2 (99.995%), Fe2O3 (⩾99.98%), WO3 (99.9%), PbO (99.99%), and TeO2 (99.99%), all received from Sigma–Aldrich. The mixtures were ground in an agate mortar then heated progressively to 650 and 900 °C for 18 h, in alumina crucibles. The resulting powders were reground and calcined at 1200 °C for 24 h in air. The samples were cooled to room temperature for re-grinding and sintering several times to improve the homogeneity.
The chemical reactions are:
 $$\eqalign{& {\rm 2SrCO}_{\rm 3} + {\rm Ca(OH)}_{\rm 2} + {\rm Fe}_{\rm 2} {\rm O}_{\rm 3} + {\rm WO}_{\rm 3} \displaystyle{{\Delta (1200\; ^\circ {\rm C})} \over {{\rm in\;} \,{\rm AIR}}} \cr & \quad \to {\rm Sr}_{\rm 2} {\rm CaFe}_{\rm 2} {\rm WO}_{\rm 9} + {\rm 2CO}_{\rm 2} + {\rm H}_{\rm 2} {\rm O};}$$
$$\eqalign{& {\rm 2SrCO}_{\rm 3} + {\rm Ca(OH)}_{\rm 2} + {\rm Fe}_{\rm 2} {\rm O}_{\rm 3} + {\rm WO}_{\rm 3} \displaystyle{{\Delta (1200\; ^\circ {\rm C})} \over {{\rm in\;} \,{\rm AIR}}} \cr & \quad \to {\rm Sr}_{\rm 2} {\rm CaFe}_{\rm 2} {\rm WO}_{\rm 9} + {\rm 2CO}_{\rm 2} + {\rm H}_{\rm 2} {\rm O};}$$ $$\eqalign{& {\rm 2SrCO}_{\rm 3} + {\rm PbO} + {\rm Fe}_{\rm 2} {\rm O}_{\rm 3} + {\rm TeO}_{\rm 2} + 1/2{\rm O}_{\rm 2} \displaystyle{{\Delta (1200\; ^\circ {\rm C})} \over {{\rm in\;} \,{\rm AIR}}} \cr & \quad \to {\rm Sr}_{\rm 2} {\rm PbFe}_{\rm 2} {\rm TeO}_{\rm 9} + {\rm 2CO}_2.}$$
$$\eqalign{& {\rm 2SrCO}_{\rm 3} + {\rm PbO} + {\rm Fe}_{\rm 2} {\rm O}_{\rm 3} + {\rm TeO}_{\rm 2} + 1/2{\rm O}_{\rm 2} \displaystyle{{\Delta (1200\; ^\circ {\rm C})} \over {{\rm in\;} \,{\rm AIR}}} \cr & \quad \to {\rm Sr}_{\rm 2} {\rm PbFe}_{\rm 2} {\rm TeO}_{\rm 9} + {\rm 2CO}_2.}$$B. X-ray powder diffraction
Diffraction data were collected at room temperature on a D2 PHASER diffractometer, with the Bragg–Brentano geometry, using Cu K α radiation (λ = 1.5418 Å) with 30 KV and 10 mA, Soller slits of 0.02 rad on incident and diffracted beams; divergence slit of 0.5°; antiscatter slit of 1°; receiving slit of 0.1 mm; with sample spinner, and a Lynxeye detector type with a maximum global count rat >1000 000 000 cps. The pattern was scanned through steps of 0.010142° (2θ), between 15 and 105° (2θ) with a fixed-time counting of 2 s/step.
The WinPlotr software (Roisnel and Rodríquez-Carvajal, Reference Roisnel and Rodríquez-Carvajal2001), integrated in the FullProf program (Rodriguez-Carvajal, Reference Rodriguez-Carvajal1990) was used to refine the crystal structure by the Rietveld method. The peak shape was described by a pseudo-Voigt function and the background level was modeled using a polynomial function.
The following structural and instrumental parameters were refined from the XRD data: scale factor, background coefficients, zero-point error, unit-cell parameters, pseudo-Voigt corrected for asymmetry parameters, Caglioti parameters (U, V, and W), atomic positions, an overall isotropic thermal factor, and antisite disorder of Fe/W and Fe/Te. The starting data needed for Rietveld refinements are the atomic positions and unit cell parameters, the model was taken from the previous studies (Ivanov et al., Reference Ivanov, Eriksson, Tellgren and Rundlof2001; Viola et al., Reference Viola, Alonso, Pedregosa and Carbonio2005).
III. RESULTS AND DISCUSSION
A. Structure determination and description of the structure
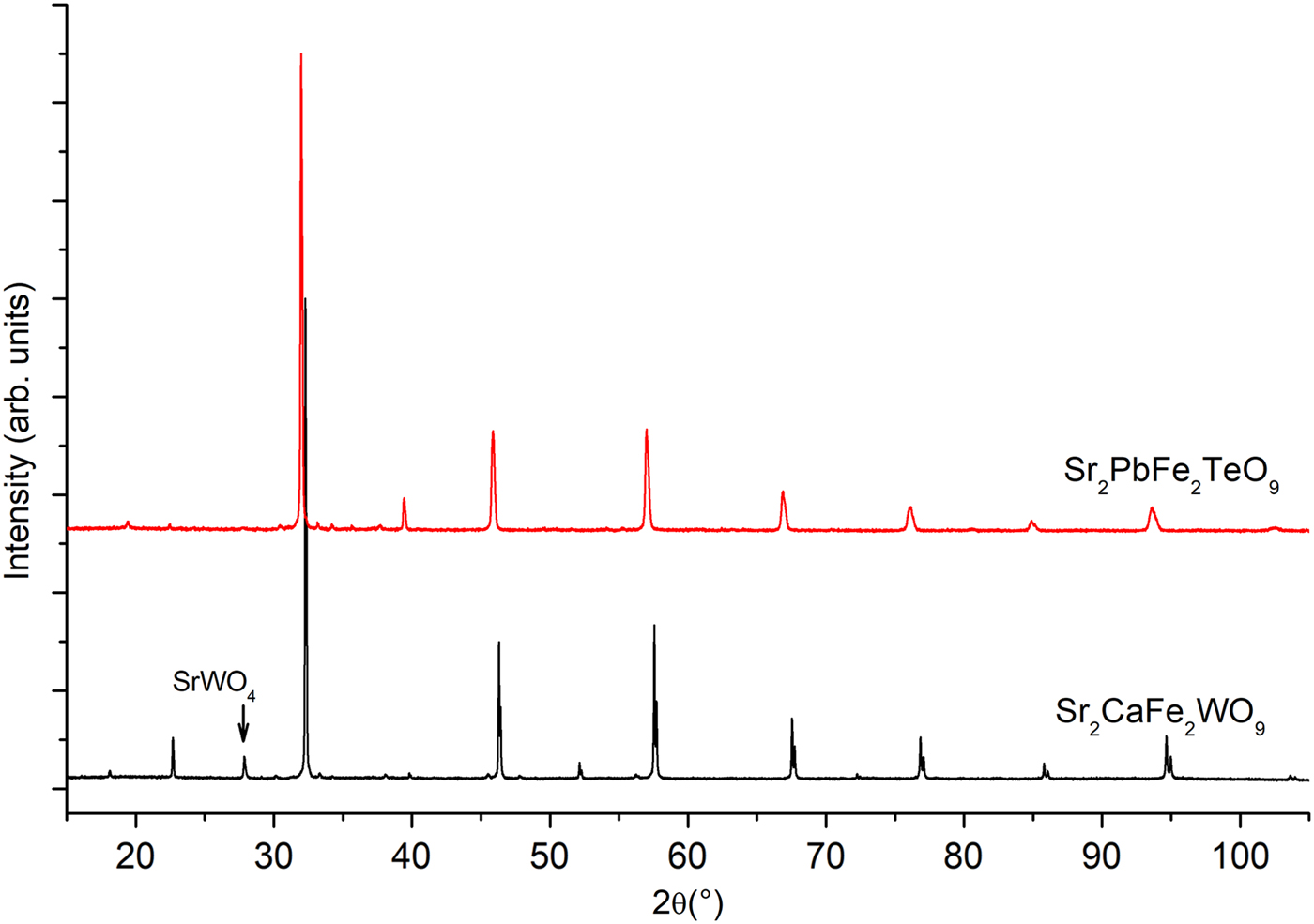
At room temperature the XRPD patterns of the compounds, with nominal compositions, Sr2CaFe2WO9 and Sr2PbFe2TeO9 are easily identified to adopt the perovskite structure (Figure 1) and by means of the computer program Dicvol (Boultif et al., Reference Boultif, Loueer and Louër1991) using the first 15 peak position with a maximal error of 0.03° as input data. The XRPD patterns were assigned to a tetragonal symmetry with I4/m as a space group for both compositions.
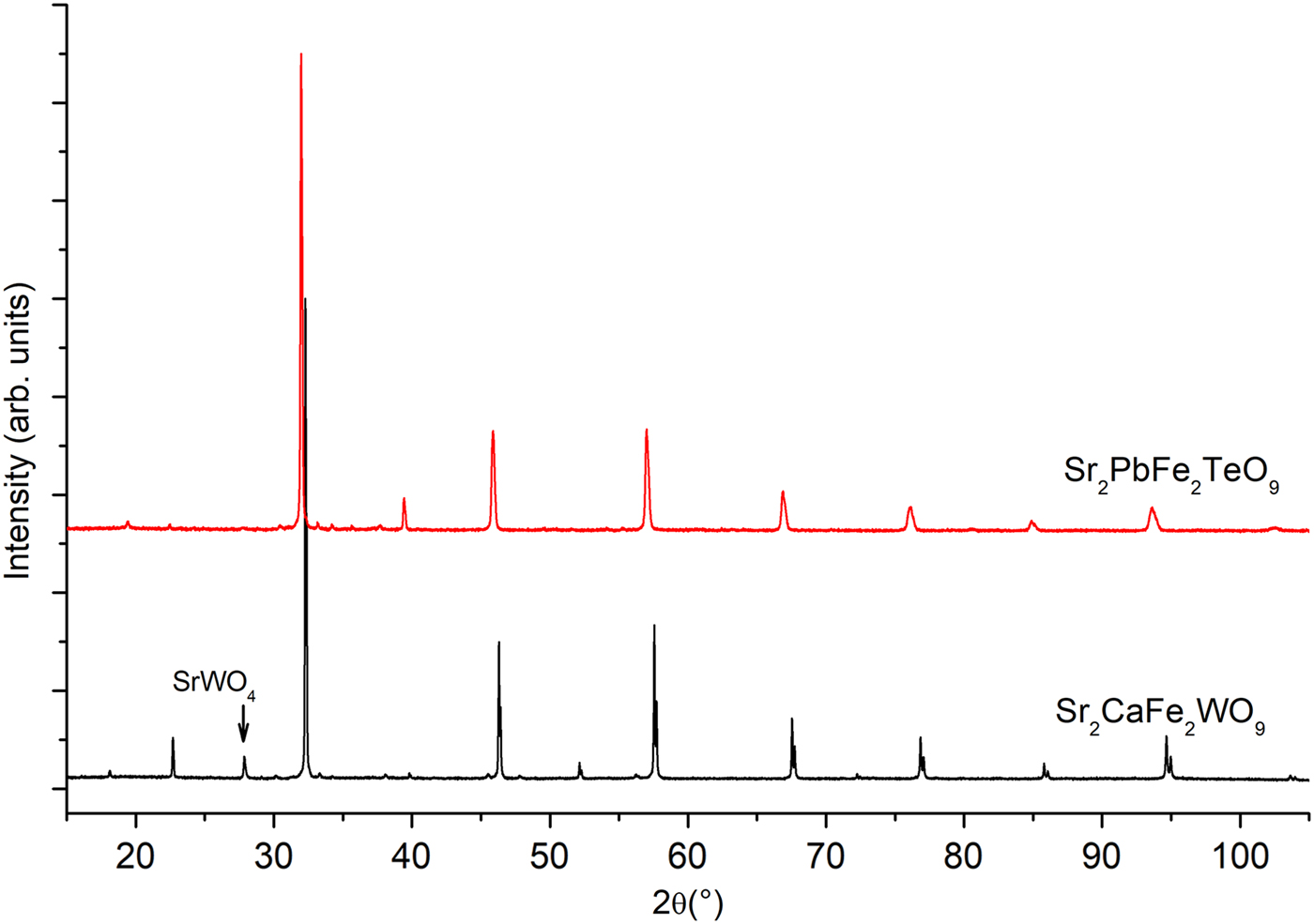
Figure 1. (Colour online) X-ray powder diffraction patterns of ceramics Sr2CaFe2WO9 and Sr2PbFe2TeO9 taken at room temperature.
Figure 1 presents the superposition of the XRPD patterns at room temperature of Sr2CaFe2WO9 and Sr2PbFe2TeO9. Bragg positions (2θ) of the peaks are shifted slightly to lower angles when we replace tungsten and calcium by tellurium and lead atoms respectively, this is may be due to electronic configuration effects and the ionic radii of those cations. The ionic radii of W6+ and Te6+ are 0.6 and 0.56 Å, respectively, and for Ca2+ and Pb2+ are 1.34 and 1.49 Å (Shannon, Reference Shannon1976). The XRPD patterns of the Sr2CaFe2WO9 and Sr2PbFe2TeO9 double perovskites were refined by Rietveld method (Rietveld, Reference Rietveld1969). The details of Rietveld refinements such as lattice parameters, cell volumes, and reliability factors are given in Table I.
Table I. Details of Rietveld refinement for Sr2CaFe2WO9 and Sr2PbFe2TeO9 double perovskites at 298 K.
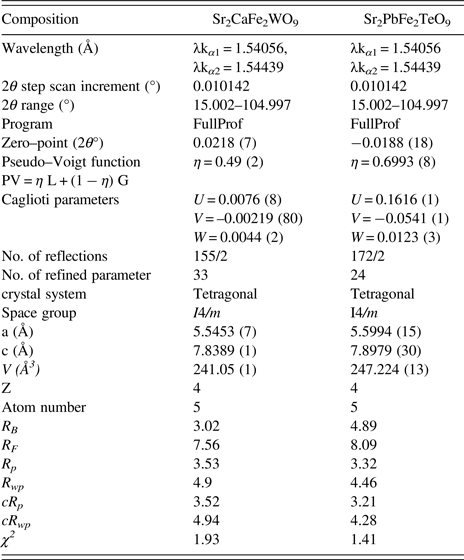
The structural refinement of Sr2CaFe2WO9 and Sr2PbFe2TeO9 taken at room temperature from XRPD data were performed in a tetragonal system with space group I4/m (ITA- No.87). Impurity corresponding to scheelite structure, SrWO4 (ICDD# 01-085-0587) was included in the refinement as the second phase with space group I41/a (ITA-No.88) for Sr2CaFe2WO9. The percentage of this impurity is about 3.7%, according to the Rietveld refinements.
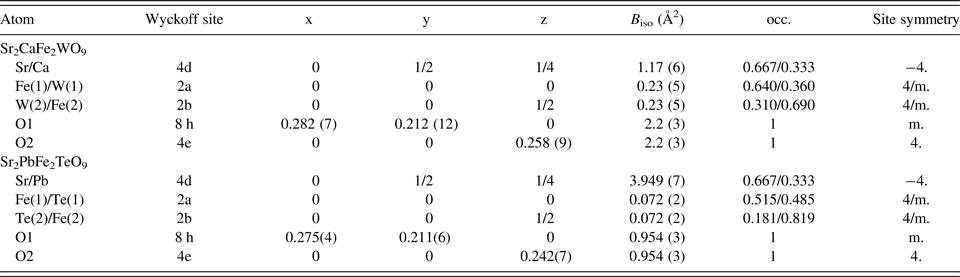
The Rietveld refinement results show that Sr, (Ca or Pb) atoms were located at the 4d (0,1/2,1/4) positions of S4 symmetry, Fe(1)/M(1) at 2a (0,0,0) and Fe(2)/M(2) at 2b (00,1/2) sites of C4h symmetry (M = W, Te), and oxygen atoms at the 8 h (x,y,0) and 4e (0,0,z) positions (Cs and C4 symmetries, respectively). The two Fe3+ and M6+ cations were found to occupy alternatively two sites; 2a and 2b with different amounts even if there are differences between the sizes and valence charges of these cations. The combination between Fe3+ and W6+ or Te6+ cations generally favors disordered arrangement in Sr2CaFe2WO9 and Sr2PbFe2TeO9. Structural details at room temperature such as atomic positions, isotropic displacement parameters, occupancy, and site symmetry are given in Table II. The Crystallographic Information Frameworks (CIF) files are available in the supplementary material.
Table II. Positional and thermal parameters for ceramics Sr2CaFe2WO9 and Sr2PbFe2TeO9 after a Rietveld refinement XRPD data collected at 298 K.

The Fe/M (M = W or Te) arrangement over the two crystallographically distinct B-sites was allowed to vary, with the constraint that the 2 : 1 composition ratio was maintained. From the structural refinement of these both compounds, an anti-site disordering effect was detected, we noted that in Sr2CaFe2WO9 some W6+ from the 2b positions randomly replace some Fe3+ at 2a positions; 2b-sites are predominantly occupied by Fe3+ atoms compared with W6+. Again, in Sr2PbFe2TeO9, some Te6+ from the 2a positions randomly replace some Fe at 2b positions; 2b-sites are predominantly occupied by Fe3+ atoms compared with Te6+. The thermal factors for (2a and 2b) positions in the space group I4/m were constrained.
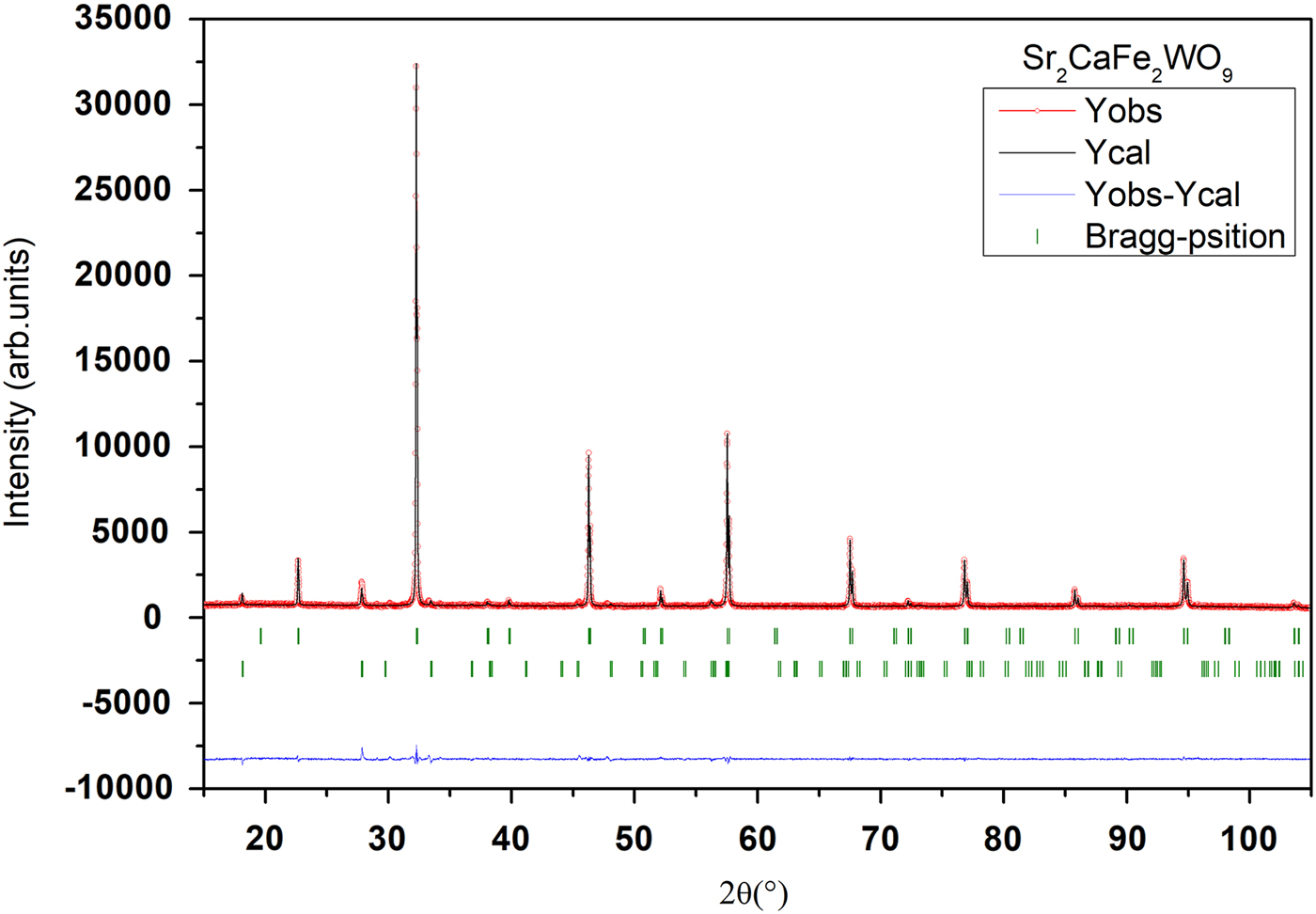
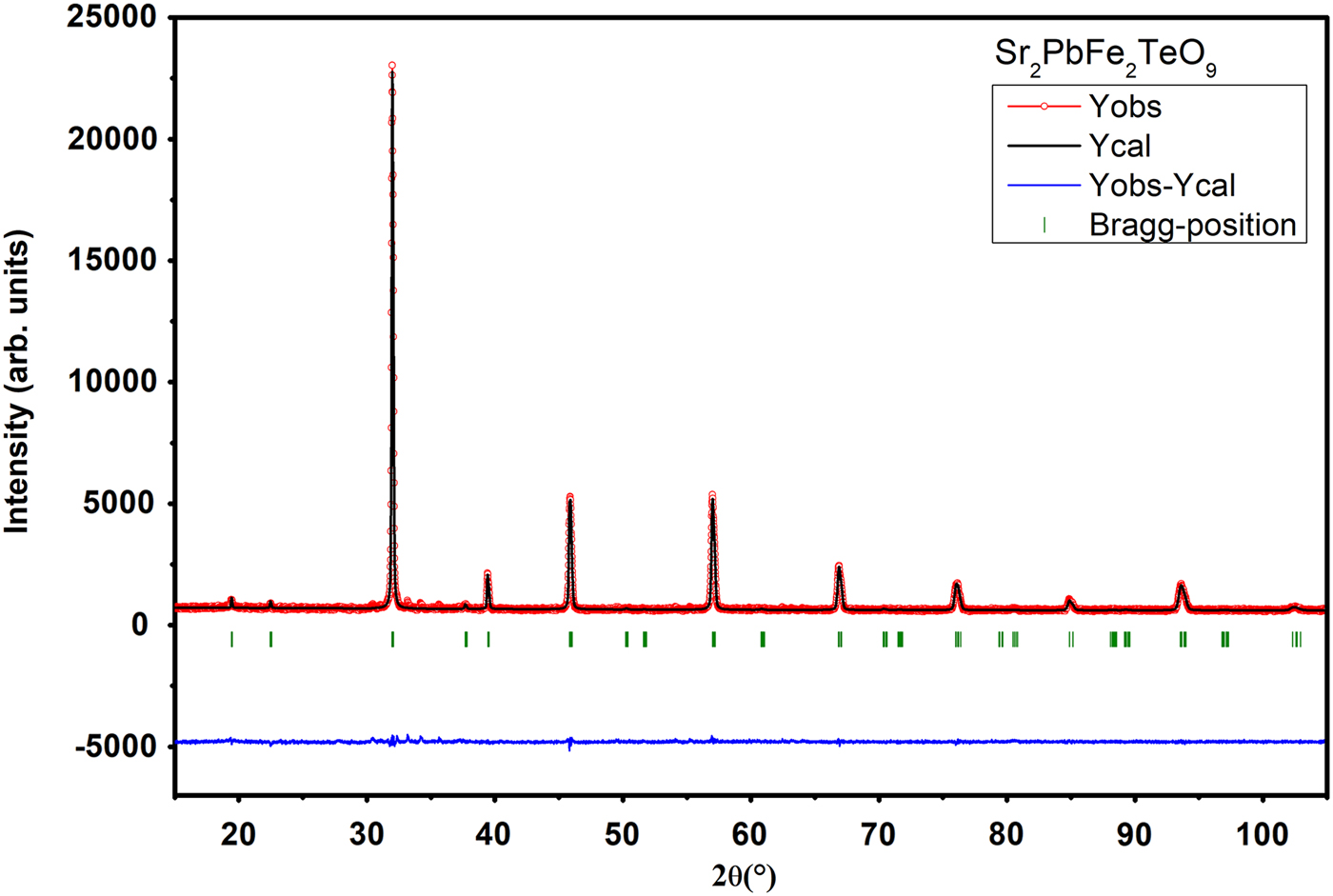
The goodness of fit is illustrated in Figures 2 and 3, showing an excellent agreement between observed and calculated XRPD patterns. The reliability factors and χ2, giving different measures of the quality of fits, are small and indicate a good quality of refinements Table I.
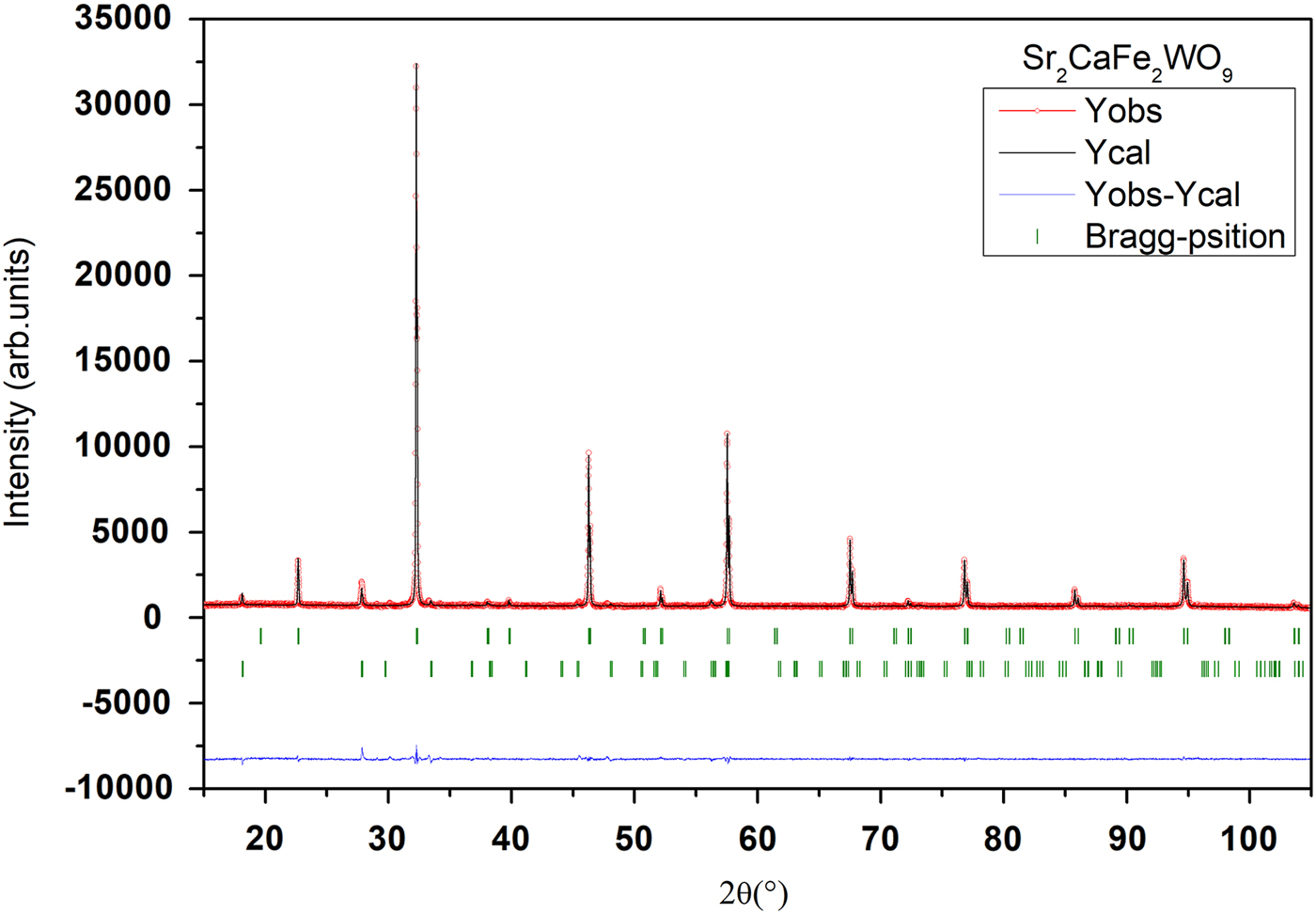
Figure 2. (Colour online) Final Rietveld plots for compound Sr2CaFe2WO9. The upper symbols illustrate the observed data (circles) and the calculated pattern (solid line). The vertical markers show calculated positions of Bragg reflections. The lower curve is the difference diagram. The second series of tick marks corresponds to the Bragg reflections of a minor impurity of SrWO4.
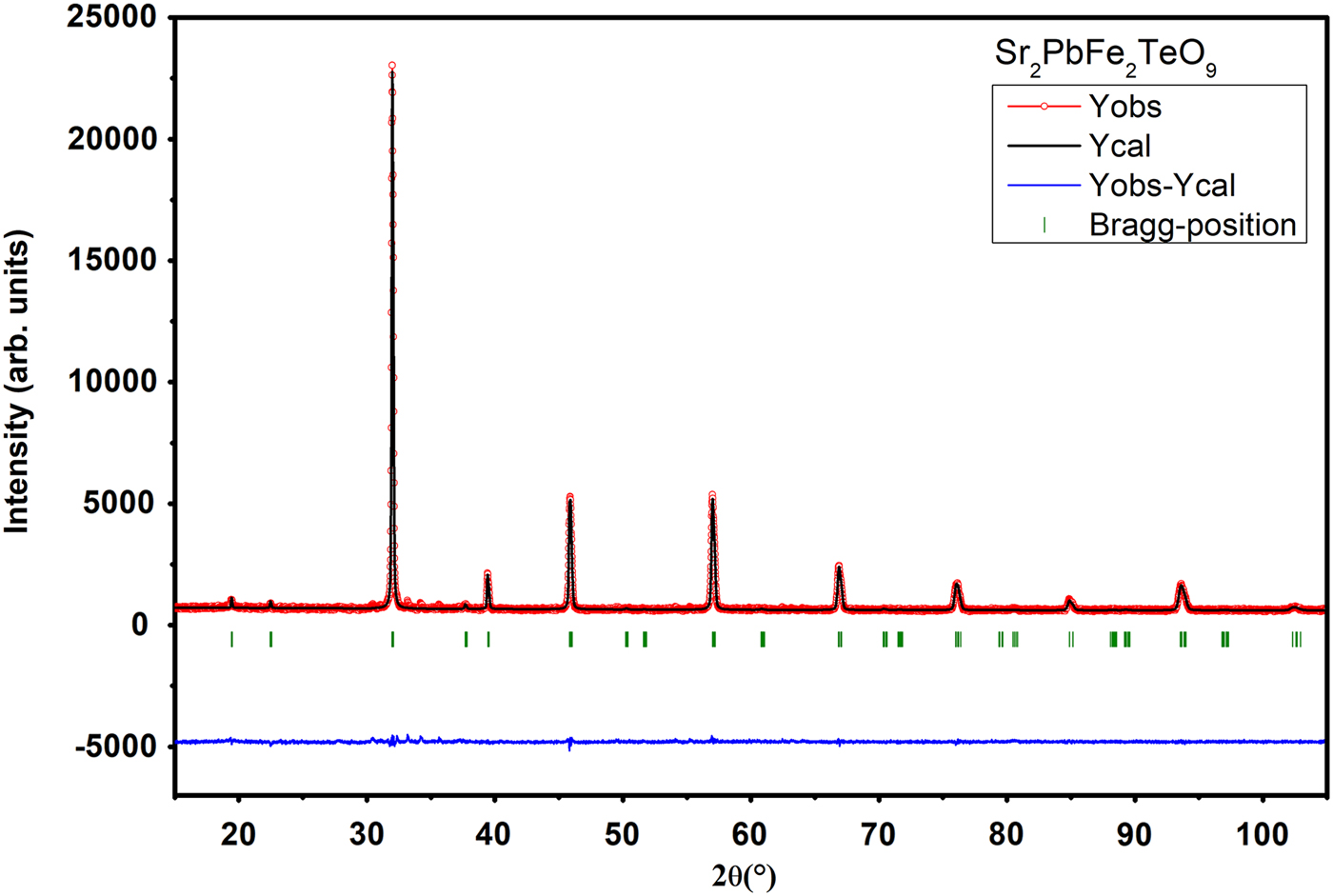
Figure 3. (Colour online) Final Rietveld plots for compound Sr2PbFe2TeO9. The upper symbols illustrate the observed data (circles) and the calculated pattern (solid line). The vertical markers show calculated positions of Bragg reflections. The lower curve is the difference diagram.
According to the crystallographic data obtained from Rietveld refinements of stoichiometric compounds Sr2CaFe2WO9 and Sr2PbFe2TeO9, Fe and W or Te cations are octahedrally coordinated with the oxygen atoms. The (Fe/W)O6/(Fe/Te)O6 octahedra are alternately connected and extended in three dimensions. Generally, the ionic sizes or electronic effects of Fe3+, Te6+, and W6+ cations have influences on the octahedral tilt angles and the associated bond angles. At room temperature, the selected bond lengths (Ǻ) and angles (°) are listed in Table III.
Table III. Selected bond lengths (Å) and angles (°) for Sr2CaFe2WO9, and Sr2PbFe2TeO9 at room temperature calculated from XRPD pattern with I4/m space group.

Structural views of Sr2CaFe2WO9 and Sr2PbFe2TeO9 is made up of alternate corner shared octahedra, i.e., (Fe/M)2aO6 and (Fe/M)2bO6 (M = W or Te). Along the c-axis, these octahedra are connected through oxygen atoms O(2); and in the ab-plane, they are connected by O(1) atoms. Due to the special positions occupied by the Fe and W or Te atoms at both 2a and 2b-sites, and oxygen atoms O(2) at 4e-sites, along the c-axis the bond angle of (Fe/M)2a–O(2)–(Fe/M)2b is constrained to be 180°. The (Fe/M)2aO6 and (Fe/M)2bO6 octahedra are ordered and alternate along the three directions, in a way that each (Fe/M)2aO6 octahedron is linked via corners to six (Fe/M)2bO6 octahedra and vice versa. The unit cell parameters were refined (Table I) with the complete powder diffraction data set (Tables IV and V). Intensities given in Tables IV and V are obtained from the “observed intensities” of the Rietveld refinement.
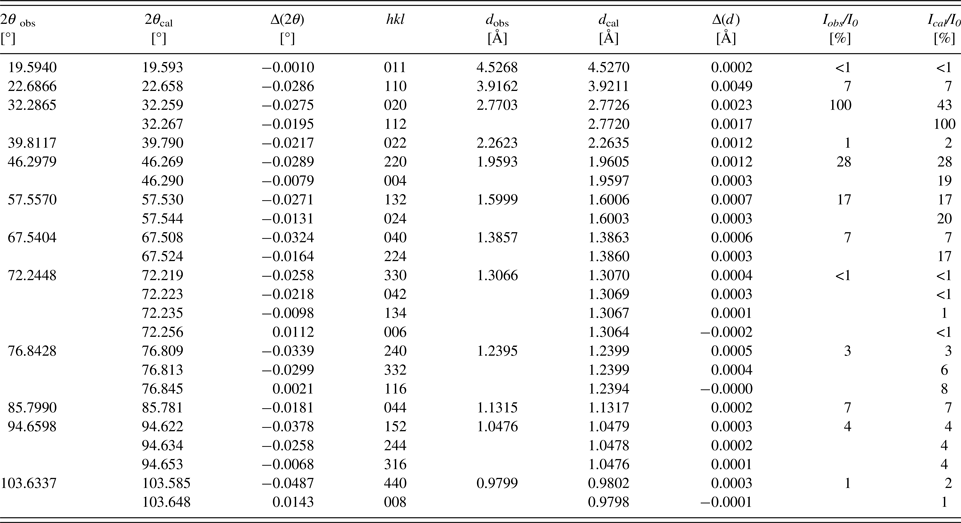
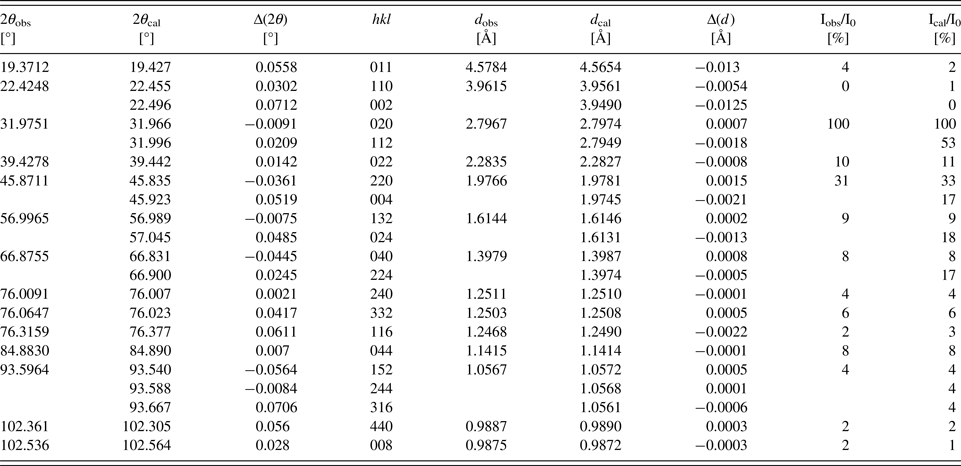
Table IV. Powder diffraction data of Sr2CaFe2WO9 (Cu K α1; λ = 1.54056 Å).

Table V. Powder diffraction data of Sr2PbFe2TeO9 (Cu K α1; λ = 1.54056 Å).
For Sr2CaFe2WO9 a drawing of the anti-phase tilting of the structure is shown in Figure 4; it contains alternating (Fe/W)2aO6 and (Fe/W)2bO6 octahedra, exhibiting a tilting angle of ψ 001 = 6.85° and tilted in anti-phase in the basal ab-plane (along the [001] direction) of the pseudo-cubic cell. This corresponds to the [a0a0c–] Glazer notation (Glazer, Reference Glazer1975) as derived byWoodward (Reference Woodward1997 for 1 : 1 ordering of double perovskites, consistent with space group I4/m. The calculated (Fe/W)2a–O(1)–(Fe/W)2b bond angle is 164.0(2)°. The (Fe/W)2aO6 octahedra exhibit an average (Fe/W)2a–O bond length of 1.98(2) Å, slightly larger than the (Fe/W)2bO6 octahedra; with an average (Fe/W)2b–O bond length of 1.968(3) Å. Along c-axis, the observed octahedra at the 2a-sites are elongated, whereas the observed octahedra at 2b-sites are significantly flattened, as shown in Table III.
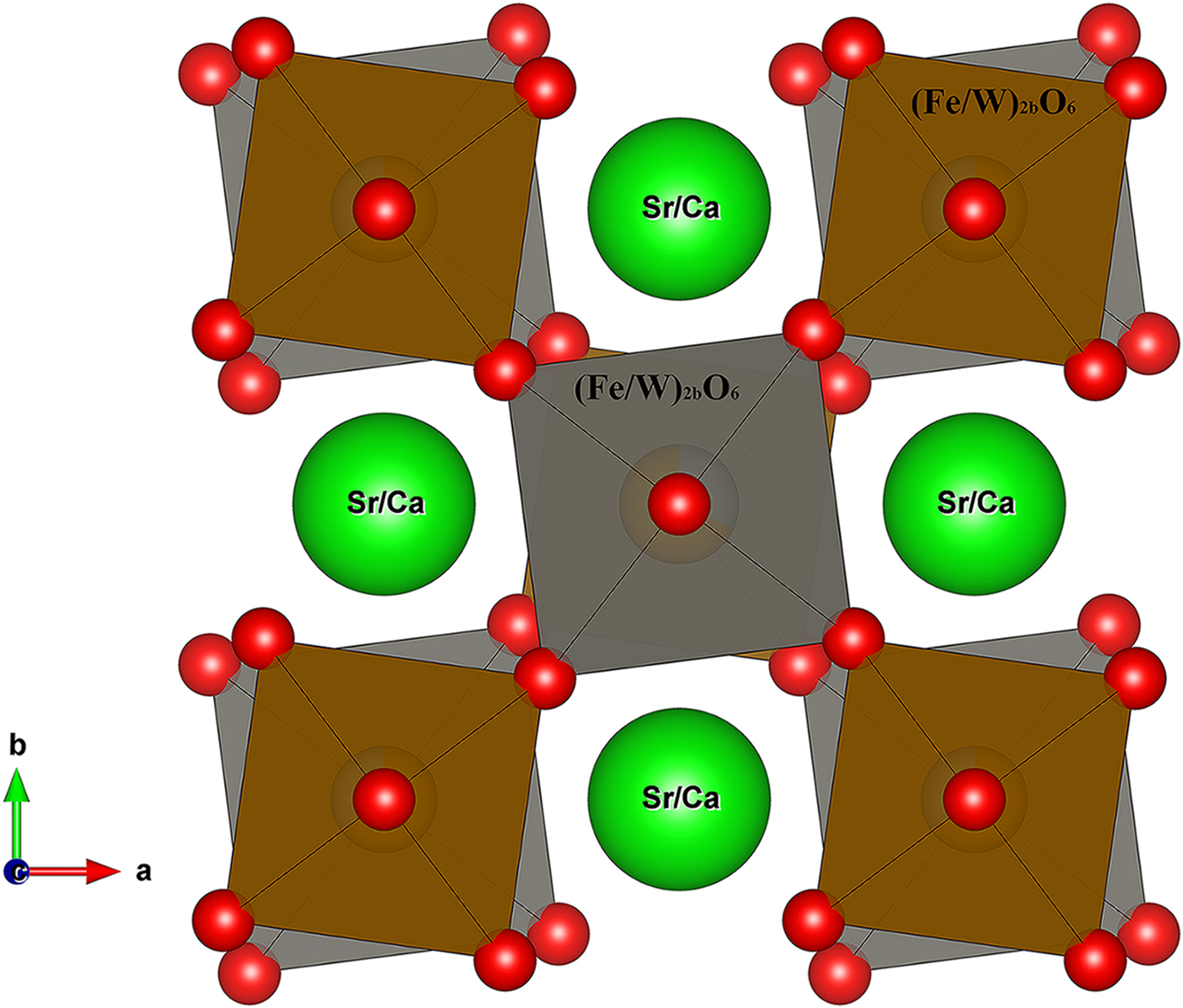
Figure 4. (Colour online) Illustrations of the tilting effect of the (Fe/W)O6 octahedra in the tetragonal structure Sr2CaFe2WO9. The octahedra are tilted in anti–phase around vertical [0 0 1] axis (Glazer notation, a0a0c–).The Fe and W cations are located inside the (Fe/W)2aO6 octahedra and (Fe/W)2bO6 octahedra.
The ionic distribution of Fe3+ and W6+ cations in Sr2CaFe2WO9 may be re-written by the formula: [Sr2Ca]4d[Fe0.962(4)W0.538(4)]2a[Fe1.038(4)W0.462(4)]2bO9, with the Fe3+/W6+ cations at the 2a-site are surrounded by six 2b-sites, which are occupied on average of 64% by Fe3+ and 36% by W6+ ions. Vice-versa, Fe3+/W6+ cations at 2b-sites are surrounded by six 2a-sites, which are occupied on average 69% by Fe3+ ions and 31% by W6+ ions.
For Sr2PbFe2TeO9 a drawing of the anti-phase tilting of the structure is illustrated in Figure 5; it contains alternating (Fe/Te)2aO6 and (Fe/Te)2bO6 octahedra, presenting a tilting angle of ψ 001 = 7.3° and tilted in anti-phase in the basal ab-plane (along the [001] direction) of the pseudocubic cell. This corresponds to the [a0a0c–] Glazer notation (Glazer, Reference Glazer1975) as derived by Woodward (Reference Woodward1997) for 1 : 1 ordering of double perovskites, consistent with space group I4/m. The calculated (Fe/Te)2a–O(1)–(Fe/Te)2b bond angle is 165.4(11)°. The (Fe/Te)2aO6 octahedra exhibit an average (Fe/Te)2a–O bond length of 1.93(2) Å, shorter than the (Fe/Te)2bO6 octahedra; with an average (Fe/Te)2b–O bond length of 2.05(3) Å. Through c-axis, the observed octahedra at the 2b –sites are elongated, whereas the observed octahedra at 2a –sites are significantly compressed, as displayed in Table III.
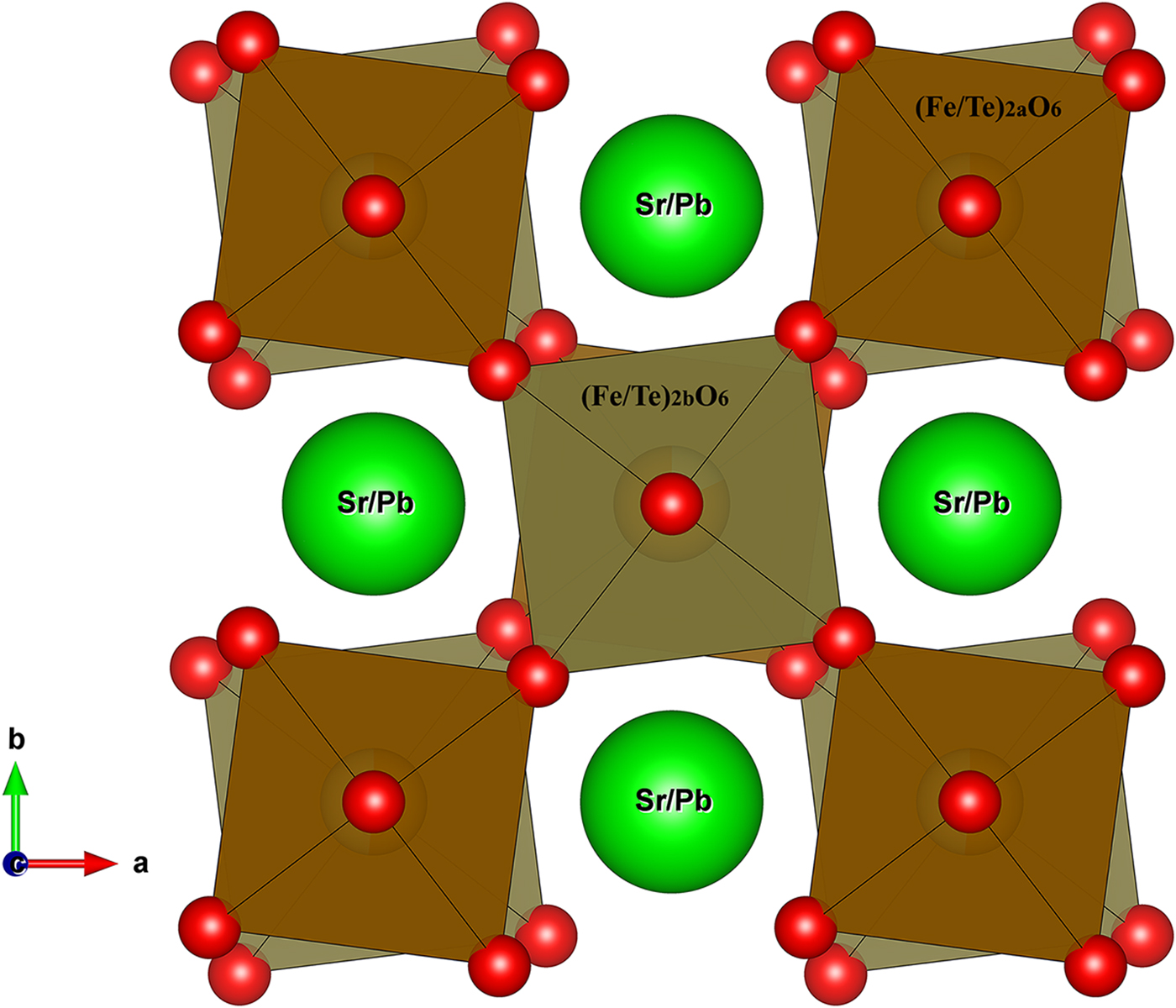
Figure 5. (Colour online) Illustrations of the tilting effect of the (Fe/Te)O6 octahedra in the tetragonal structureSr2PbFe2TeO9. The octahedra are tilted in anti–phase around vertical [0 0 1] axis (Glazer notation, a0a0c–).The Fe and Te cations are located inside the (Fe/Te)2aO6 octahedra and (Fe/Te)2bO6 octahedra.
The ionic distribution of Fe3+ and Te6+ cations in Sr2PbFe2TeO9 perhaps re–written by the formula: [Sr2Pb]4d[Fe0.772(2)Te0.728(2)]2a[Fe1.228(2)Te0.272(2)]2bO9, with the Fe3+/Te6+ cations at the 2a-site are surrounded by six 2b-sites, which are occupied on average 51.5% by Fe3+ and 48.5% by Te6+ ions. Fe3+/Te6+ cations at 2b-site are surrounded by six 2a-sites, which are occupied on average 81.9% by Fe3+ ions and 18.1% by Te6+ ions.
In fact, the average (Fe/M)–O bond lengths obtained from Rietveld refinements of the compounds Sr2CaFe2WO9 and Sr2PbFe2TeO9 are very similar to those found in other oxides with Fe3+ and W6+ or Te6+ cations (Ivanov et al., Reference Ivanov, Eriksson, Tellgren and Rundlof2001; Viola et al., Reference Viola, Alonso, Pedregosa and Carbonio2005; Manoun et al., Reference Manoun, Benmokhtar, Bih, Azrour, Ezzahi, Ider, Azdouz, Annersten and Lazor2011) and in agreement with the individual Fe3+, W6+, Te6+–O bond length calculated from Shannon's ionic radii (Fe–O = 2.065 Ǻ, W–O = 2.02 Ǻ and Te–O = 1.96 Å) for high-spin electron configuration in octahedral coordination (Shannon, Reference Shannon1976).
Antisite-disorder of Fe+3 and W+6 or Te+6 cations on B and B′ sites in the perovskite structure with general formula A3B2B′O9 or A2BB′O6, has a direct effect on the physical properties (Baum et al., Reference Baum, Stewart, Mercader and Grenèche2004; Ivanov et al., Reference Ivanov, Eriksson, Erikssen, Tellgren and Rundlof2004; Pinacca et al., Reference Pinacca, Viola, Alonso, Pedregosa and Carbonio2005). In our case the ordering of iron and tungsten/tellurium cations is located in 2a and 2b sites of the tetragonal structure (I4/m). The formula, for Sr2CaFe2WO9, can be written as: [Sr2Ca]4d[Fe0.962(4)W0.538(4)]2a[Fe1.038(4)W0.462(4)]2bO9 or [Sr0.667Ca0.333]4d[Fe0.641W0.359]2a[Fe0.69W0.31]2bO6, and for Sr2PbFe2TeO9 it can be written as [Sr2Pb]4d[Fe0.772(2)Te0.728(2)]2a[Fe1.228(2)Te0.272(2)]2bO9 or [Sr0.667Pb0.333]4d[Fe0.515Te0.485]2a[Fe0.819Te0.181]2bO6.
Previously Ivanov et al. (Reference Ivanov, Nordblad, Eriksson, Tellgren and Rundlöf2007), reported on the magnetoelectric perovskite Sr3Fe2TeO9 using magnetic measurements and neutron powder diffraction, it was shown that the antisite-disorder behavior has a direct effect on the magnetic property. Correlation between the structural and magnetic properties was also reported, the magnetic temperature transition (TN) is strongly related to the value of the Fe3+ – O – Te6+ angle and with lattice distortions. Similar works are also reported for Sr3Fe2TeO9, Sr3Fe2MoO9, Sr3Fe2ReO9, and Sr3Fe2UO9 magnetic compounds. Hence, the antisite-disorder which is independent of bond length distortion can be controlled by calculating the relative deviation (relative distortion) from average bond length of the polyhedra Δoct given by Fleet (Reference Fleet1976)
 $$\Delta _{oct} = \displaystyle{1 \over 6}\sum\limits_{i = 1}^n {\left[ {(l_i - l_0 )/l_0} \right]^2}. $$
$$\Delta _{oct} = \displaystyle{1 \over 6}\sum\limits_{i = 1}^n {\left[ {(l_i - l_0 )/l_0} \right]^2}. $$where l 0 is the theoretical bond length calculated from the ionic radii taken from Shannon tables (Shannon, Reference Shannon1976), and l i is the bond length obtained from the Rietveld refinement (see Table III). The relative distortion of (Sr/Ca)4dO12-polyhedra, (Fe/W)2aO6 and (Fe/W)2bO6-octahydra, are illustrated in Table VI. Also, the relative deviation for Sr3Fe2WO9 and Sr3Fe2TeO9 are given for comparison.
Table VI. Calculated relative deviation from average bond length of the polyhedra of Sr2CaFe2WO9, Sr2PbFe2TeO9, Sr3Fe2WO9, and Sr3Fe2TeO9.

a Crystallographic information of Sr3Fe2WO9 and Sr3Fe2TeO9 are taken from refs (Ivanov et al., Reference Ivanov, Eriksson, Tellgren and Rundlof2001) and (Ivanov et al., Reference Ivanov, Nordblad, Eriksson, Tellgren and Rundlöf2007) for comparison.
IV. CONCLUSION
In this paper, using XRPD technique, we reported on the crystal structure of Sr2CaFe2WO9 and Sr2PbFe2TeO9 double perovskite oxides. The tetragonal system with space group I4/m was adopted for both compounds. The lattice parameters and volumes were different; this is due to larger size of Pb2+ compared with that of Ca2+. The Fe and W and/or Te cations are partially disordered over the B-sites, indicating the presence of a partial amount of W and/or Te at Fe positions and vice versa.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0885715618000222
Acknowledgements
The authors are grateful to the Office Chérifien des Phosphates in the Moroccan Kingdom (OCP group) and Mohammed VI Polytechnic University, the University Hassan 1st for its support and the Swedish Research Council for the financial grant SRL(MENA) # 348- 2014-4287.