Significant outcomes
∙ In non-depressed people, elevated alpha power during non-rapid eye movement (NREM) sleep and a low delta sleep ratio (DSR) both predict subsequent increases in depression symptoms during interferon-alpha (IFN-α) therapy.
∙ These quantitative measures of electroencephalogram (EEG) power are both highly correlated between nights.
Limitations
∙ The sample size is fairly small, and serial EEG measures were not obtained during IFN-α therapy.
∙ The results may be specific for depression occurring during IFN-α therapy, and generalisation to other types of depression is unknown.
Introduction
The incidence of a major depressive disorder (MDD) episode is usually <2%/year (Reference Patten1,Reference Eaton, Kramer, Anthony, Dryman, Shapiro and Locke2), even in medically ill populations. An MDD episode occurs in <5–10% of people over a several year period (Reference Patten and Lee3,Reference Kruijt, Antypa and Booij4), and in only about 12% the first year after a cancer diagnosis (Reference Boyes, Girgis, D'Este, Zucca, Lecathelinais and Carey5). Treatment with high doses of IFN-α results in an incidence of about 25% in the first few months (Reference Udina, Castellvi and Moreno-Espana6). In each of these situations, most people are resilient – with only a minority exhibiting vulnerability to the development of depression. As depression is a major cause of disability and premature death, replicating this resilience in vulnerable people and thereby preventing depression would be of great consequence.
Because poor sleep frequently precedes the future development of MDD (Reference Breslau, Roth, Rosenthal and Andreski7–Reference Roane and Taylor10), it may be possible to prevent depression by correcting indices of sleep. Nonetheless, it is not clear which sleep abnormality might represent a pre-existing vulnerability to subsequent MDD. Cross-sectional studies find a number of objective sleep parameters that are associated with MDD including sleep continuity disturbances (prolonged sleep latency, nocturnal arousals, and early morning awakening), diminished slow wave sleep (SWS), shortened rapid eye movement (REM) latency, decreased DSR, among others (Reference Emslie, Armitage, Weinberg, Rush, Mayes and Hoffmann11–Reference Wichniak, Wierzbicka and Jernajczyk17). However, to our knowledge there are no studies that have examined which of these parameters precede or antedate MDD.
To prospectively address which aspect of ‘poor’ sleep precedes depression, we have therefore employed quantitative polysomnography (PSG) in non-depressed, euthymic individuals who are intending to start IFN-α therapy. IFN-α treatment-related MDD has an incidence of about 25% in the first few months (Reference Udina, Castellvi and Moreno-Espana6,Reference Felger and Lotrich18), thereby permitting examination of subjects before they start IFN-α treatment – and before they develop MDD. Consistent with other instances of MDD, poor sleep quality before IFN-α therapy strongly predicts risk for developing MDD during IFN-α treatment (Reference Franzen, Buysse, Rabinovitz, Pollock and Lotrich19), even when controlling for baseline depression and/or history of prior depression. In time-lagged hierarchical regression during IFN-α therapy, subjectively poor sleep is a strong predictor of increased Beck Depression Inventory (BDI) scores 1 month later though not vice versa (Reference Prather, Rabinvitz, Pollock and Lotrich20), further demonstrating that poor self-reported sleep quality precedes depression in this population.
We focused our initial attention on delta power (0.5–4 Hz) during NREM sleep. Delta power is an index of the restorative function of sleep (Reference Campbell, Darchia and Higgins21–Reference Dworak, McCarley, Kim, Kalinchuk and Basheer23). Delta waves propagate across the cortex during sleep, and consist of large currents in the medial frontal gyrus, the inferior frontal gyrus, the anterior cingulate, the precuneus, and the posterior cingulate (Reference Murphy, Riedner, Huber, Massimini, Ferrarelli and Tononi24). In positron emission tomography studies, delta power during NREM sleep is highly correlated with decreased metabolic activity in the ventromedial prefrontal cortex, anterior cingulate, and orbitofrontal regions (Reference Dang-Vu, Desseilles and Laureys25,Reference Nofzinger26) – regions implicated in MDD. Conversely, delta power is diminished during periods of elevated stress (Reference Hall, Thayer and Germain27) and the normally reduced metabolic activity during NREM sleep is likewise abnormal during depression (Reference Germain, Nofzinger, Kupfer and Buysse28). It is therefore plausible that disrupting NREM-related restorative processes increases vulnerability to subsequent MDD precipitating insults.
The majority of restorative delta power normally occurs during the first NREM period, with exponentially decreasing delta power in subsequent NREM periods. A basic index of this decrease in delta power over the night is the ratio of delta waves during the first NREM period to the second NREM period – the DSR. A lower DSR is consistently present in MDD, where DSR is about 1.6 in normal subjects compared with 1.1 in depressed subjects (Reference Armitage, Hoffmann, Trivedi and Rush13,Reference Kupfer, Reynolds, Ulrich and Grochocinski29). Importantly, DSR in euthymic individuals is a robust predictor of subsequent MDD relapse after cognitive therapy (Reference Kupfer, Frank, McEachran and Grochocinski16,Reference Spanier, Frank, McEachran, Grochocinski and Kupfer30), after interpersonal psychotherapy (Reference Buysse, Frank, Lowe, Cherry and Kupfer31), and/or during antidepressant maintenance therapy (Reference Reynolds, Buysse and Brunner32). Because low DSR has been replicated as a risk for depression relapse, our primary hypothesis was that low DSR would be associated with risk for inflammation-related depression.
Another aspect of MDD that has been repeatedly observed is shortened REM latency (Reference Steiger and Kimura14,Reference Kupfer, Reynolds, Ulrich and Grochocinski29,Reference Lauer, Schreiber, Holsboer and Krieg33). The pressure for REM to occur earlier may shorten the duration of the initial NREM period (Reference Buysse, Hall and Tu12), and thereby affect DSR. Finally, quantitative EEGs (qEEGs) were used to determine if the per cent of activity in particular frequencies would be associated with depression risk. In addition to delta power, we examined the predictive roles of elevated alpha power (Reference Fenzl, Touma and Romanowski34,Reference Nitschke, Heller, Etienne and Miller35), as well as theta (Reference von Stein and Sarnthein36), sigma, and beta power (Reference Bjorvatn, Fagerland and Ursin37). Elevations in some higher frequencies may reflect, in part, hyperarousal during sleep (Reference Nofzinger, Price and Meltzer38,Reference Krystal and Edinger39). Intrusion of these other frequencies could also adversely affect delta power.
The aspect of sleep that antedates development of MDD has consequences for prevention strategies. Different insomnia treatments typically have different effects on sleep parameters. As examples, benzodiazepines may improve sleep latency but can decrease delta power and increase beta power (Reference Bastien, LeBlanc, Carrier and Morin40,Reference Feinberg, Maloney and Campbell41); serotonin reuptake inhibitors suppress REM sleep (Reference Dumont, de Visser, Cohen and van Gerven42,Reference Silvestri, Pace-Schott, Gersh, Stickgold, Salzman and Hobson43); mindfulness meditation improves sleep continuity but can decrease SWS (Reference Britton, Haynes, Fridel and Bootzin44); zolpidem decreases alpha power with limited effects on SWS while gaboxadol improves delta power (Reference Lundahl, Deacon, Maurice and Staner45); and agomelatine improves sleep latency with minimal effect on REM latency, while escitalopram improves REM latency with minimal effect on sleep latency (Reference Quera-Salva, Hajak and Philip46). There are a large variety of interventions available to improve sleep. Which of these multiple and various sleep interventions affect parameters that precede depression and could therefore be examined for prevention of depression?
Methods
All procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008, and as approved by the University of Pittsburgh Institutional Review Board. A total of 24 non-depressed adult subjects with chronic hepatitis C were screened to not have active mood disorders (currently or in the past 6 months), psychotic disorders, or substance abuse, using the Structured Clinical Interview for DSM-IV (SCID-IV) (Reference First, Spitzer and Williams47). The SCID-IV was performed by a psychiatrist or by a trained research assistant and then reviewed with the psychiatrist. When a urine drug screen was available, this was used to confirm SCID-IV findings. Importantly, use of any hypnotic agents was a reason for exclusion. Exclusion criteria also included known obstructive sleep apnoea, active inflammatory illness requiring corticosteroid treatment, endocrinopathy (such as hypothyroidism), neurologic disorder such as tumour or epilepsy, and pregnancy (or other medical contra-indication to IFN-α therapy). In this highly screened group, subjects had an average cumulate illness rating scale scores <4 (Table 1) consistent with few medical co-morbidities other than hepatitis C virus (HCV).
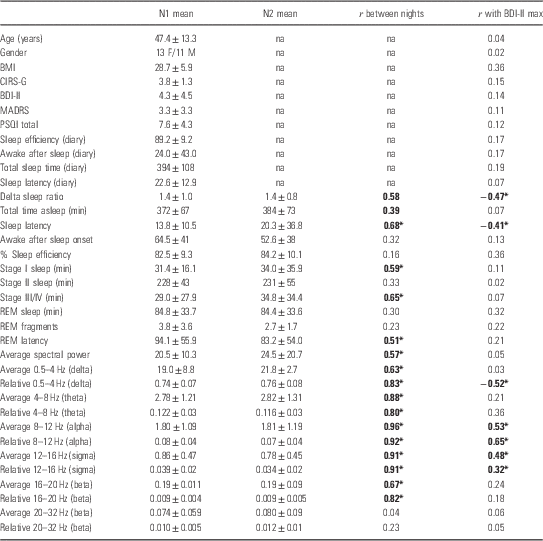
Table 1 Demographics and polysomnography results include the body mass index (BMI), cumulative illness rating scale for geriatrics (CIRS-G), Beck Depression Inventory-II (BDI-II), Montgomery-Asberg Depression Rating Scale (MADRS), diary self-reports of sleep, and quantitative EEG results

PSQI, Pittsburgh Sleep Quality Index.
The linear correlation coefficient (R) with maximal change in BDI-II scores is given (bold with asterisk indicates p<0.05).
Baseline BDI-II (Reference Beck, Steer and Garbin48) scores were obtained before starting IFN-α therapy, and then every 2 weeks after starting therapy. Depression vulnerability was quantified as the maximal increase in BDI-II score for each individual during treatment. Because subjects could be started on antidepressant treatment if they developed a major depressive episode, the maximal change in BDI-II provides a quantitative measure that is minimally confounded by subsequent alleviation of depression symptoms by medication. Participants also completed the Pittsburgh Sleep Quality Index (PSQI) (Reference Buysse, Reynolds, Monk, Berman and Kupfer49). The PSQI is a well-validated 18-item self-report measure of global sleep quality. Scores below 5 reflect good sleep quality (Reference Hayashino, Yamazaki, Takegami, Nakayama, Sokejima and Fukuhara50), but a cut-off >7 has been proposed to indicate poor sleep in chronically ill samples (Reference Owen, Parker and McGuire51,Reference Carpenter and Andrykowski52).
A 2-week sleep diary (Reference Monk, Reynolds and Kupfer53), to be completed each morning upon awakening, was requested from subjects before PSG. The diary provided prospective measures of sleep latency, wake time after sleep onset, total sleep duration, and sleep efficiency (i.e. ratio of total sleep duration/total time spent in bed). All subjects then completed a first night of PSG before IFN-α therapy to screen for sleep disorders such as sleep apnoea, and to provide habituation to the PSG environment. PSG was performed at the Western Psychiatric Institute and Clinic using GrassTelefactor M15 bipolar Neurodata amplifiers and locally developed collection software (Reference Doman, Detka and Hoffman54). Exclusion criteria included an apnoea-hypopnea index >15 and/or index of periodic leg movements with arousal (PLMA-I) >15. After this initial screening night and after at least one intervening night at home, a second PSG night was conducted. The recording montage consisted of bilateral central EEG leads referenced to A1+A2; right and left electro-oculogram referenced to A1+A2; and bipolar submentalis electromyogram. EEG data were acquired at a rate of 256 Hz and decimated to 128 Hz. High frequency EEG artefacts were excluded in 4-s bins with a previously validated algorithm (Reference Brunner, Vasko, Detka, Monahan, Reynolds and Kupfer55). EEG spectra for each artefact-free 4-s epoch were then aligned with 20-s visually scored sleep stage data to exclude epochs scored as awake or REM sleep. Scoring was performed by trained PSG technicians who maintained a high level of scoring reliability, as indicated by mean κ values of >0.80 for various sleep stages, use standard criteria and Stellate Harmonie software.
qEEG power values from artefact-free 4-s epochs were averaged over NREM periods in 0.5 Hz bins before modelling and analysis. DSR was operationalised as the average number of delta counts per minute in the first NREM period divided by the number of delta counts per minute in the second period (Reference Kupfer, Frank, McEachran and Grochocinski16). Other sleep variables included average and relative power at various frequencies [0.5–4 Hz (delta), 4–8 Hz (theta), 8–12 Hz (alpha), 12–16 Hz (sigma), and 20–32 Hz (beta)], total sleep duration (from diary self-report and from PSG), sleep latency (from diary and from time from beginning of the recording period to the first of 10 consecutive minutes of stage 2 or stages 3–4 sleep interrupted by no more than 2 min of stage 1 or wakefulness), sleep efficiency (time spent asleep/total recording period×100), percentage of time in stages 1, 2, and SWS (sum of sleep stages 3 and 4), REM latency (time between sleep onset and the first REM period with ≥3 consecutive minutes of REM sleep), average REM counts, and REM density.
To avoid confounding, the sleep question was excluded from the BDI-II total scores used in the analyses. Using SPSS 20.0, we used linear regression to examine the correlation of the maximal change in BDI-II with individual sleep variables. To ensure the validity of these regressions, we ensured that the standardised residuals were normally distributed. The role of DSR was our primary hypothesis, with the other sleep variables being exploratory (statistical corrections for repeated testing were not applied). Subsequent repeated-measure exploratory analyses of BDI-II scores employed mixed-effect analyses with unstructured covariances. Results are presented as mean±standard deviation.
Results
Subjects were euthymic (not depressed) before starting IFN-α therapy as evidenced in both BDI-II and MADRS scores (Table 1), although overall self-reported sleep quality was typically poor (average PSQI>7). This was similar to prior studies of hepatitis patients (Reference Franzen, Buysse, Rabinovitz, Pollock and Lotrich19,Reference Prather, Rabinvitz, Pollock and Lotrich20). Sleep efficiency based on diary self-reports was similar to that calculated by PSG observations; and spectral power was remarkably similar across the two nights of testing (Table 1). For subsequent analyses, the two nights were therefore averaged. Typical in this population, 25% had a past history of a major depressive episode in remission; 45% had a history of drug abuse/dependence in remission; and 45% had a history of alcohol abuse/dependence in remission. The average time of abstinent remission was 9.5 months. One subject also had a history of panic disorder in remission, and one had a history of post-traumatic stress disorder in remission. None had a history of bipolar disorder, psychotic disorder, or obsessive-compulsive disorder.
Excluding the sleep question, BDI-II increased during IFN-α therapy with a maximal change that averaged 9.4±6.8 (range: −1 to 23). The maximal increase in BDI-II was not associated with a history of prior MDD (p=0.74), past alcohol abuse/dependence (p=0.34), nor past drug abuse/dependence (p=0.94). Diary self-report measures did not correlate with subsequent maximal change in BDI-II, nor did sleep efficiency or minutes spent in the various stages of sleep (Table 1). REM latency also was not predictive of subsequent depression (Table 1). However, as hypothesised, pre-treatment DSR was negatively correlated with the maximal increase in BDI-II during INF-α treatment (B=−0.59±0.025; t=−2.3; p=0.03), as was lower relative delta power overall (Table 1). Interestingly, sleep latency was also negatively correlated with maximal BDI-II increase, indicating that possibly a greater sleep drive and need for sleep (and therefore falling asleep more quickly) was predictive of future depression. In subsequent exploration of the power spectral results, elevated relative alpha and relative sigma power (but not higher beta frequencies) were also predictive of increased BDI-II (Table 1). In this exploration, Bonferroni correction was not performed, but for 35 independent tests would require p<0.0014.
DSR correlated with several other sleep variables including REM latency (r 2=0.42; p=0.009), alpha power (r 2=0.47; p<0.0001) and sigma power (r 2=0.34; p=0.003). Because of these multiple inter-correlations among sleep parameters, each of the sleep variables that were associated with maximal BDI-II increase was therefore included in stepwise (forward and backward) regression analyses. The only variable that remained associated with maximal change in BDI-II in the stepwise analyses was relative alpha power, where r 2=0.38; p<0.002 (Fig. 1). The pattern was similar for sigma power, but it was no longer significantly associated in stepwise regression. DSR also lost significance when including alpha power in the model.
Fig. 1 Pre-treatment alpha power is predictive of subsequently increased Beck Depression Inventory-II (BDI-II) scores.
To further explore the relationship between alpha power and DSR on depression vulnerability, we next dichotomised both sleep parameters (using a median split for each) and used both in repeated measure mixed-effect analysis of BDI-II scores. Both DSR and max-change in BDI-II were normally distributed (Kolmogorov–Smirnov test), but alpha power was not. We focused on the first couple months of IFN-α therapy before any antidepressant interventions or treatment interruption could confound the data. Both DSR and alpha power were associated with increasing BDI-II over time (p<0.001 for each). Importantly, there was a significant interaction between time, DSR, and alpha power [F(13,511)=228; p<0.0001]. As seen in Fig. 2, subjects are resilient to depression when both DSR and alpha power are both low. However, when alpha power was high, then subjects developed depression regardless of DSR. Results were similar when using log-transformed alpha power.
Fig. 2 When relative alpha power is low (upper panel), then a high delta sleep ratio (DSR; filled circles) is protective against depression compared with low DSR (open circles). When relative alpha power is high (lower panel), then depression worsens regardless of DSR.
Discussion
Only a few sleep parameters in non-depressed individuals were associated with future risk for increased depression symptoms during IFN-α therapy. Neither sleep efficiency nor REM latency were associated with future risk for depression. Total sleep time or the time spent in any particular stage of sleep was also not predictive. But we confirmed our primary hypothesis that low DSR would predict worsening depression, and additionally observed that relative power in the 8–16 Hz range was also predictive. Thus, standard PSG was not able to predict vulnerability to IFN-α, but qEEG was useful in this regard.
DSR is believed to index the homeostatic drive and restorative function of delta sleep (Reference Campbell, Darchia and Higgins21–Reference Dworak, McCarley, Kim, Kalinchuk and Basheer23). Normally, this homeostatic sleep drive should exponentially dissipate through the night, with decreasing delta power in each successive NREM period. Conversely, both alpha and sigma power are considered to be markers of hyperarousal during sleep and can be elevated during depression (Reference Lange56). Alpha activity is associated with arousal-like states that are more easily interrupted by noise – and potentially worse subjective sleep quality (Reference McKinney, Dang-Vu, Buxton, Solet and Ellenbogen57). Highly stress-reactive mice have elevated alpha activity (Reference Fenzl, Touma and Romanowski34), as do children with family histories of alcohol abuse (Reference Dahl, Williamson, Bertocci, Stolz, Ryan and Ehlers58). Consistent with this, patients with gastroesophageal reflux disease have lower delta and higher alpha activity (Reference Budhiraja, Quan, Punjabi, Drake, Dickman and Fass59). Likewise in patients with back pain, more central alpha activity is noted in depressed patients (Reference Harman, Pivik, D'Eon, Wilson, Swenson and Matsunaga60).
Related to the role of hyperarousal, decreased activation of the reticular formation may be necessary for hyperpolarisation of thalamocortical neurons, decreased sensory input to the cortex, and the development of delta waves (Reference Steriade and McCarley61). Stress, potentially contributing to hyperarousal, can increase alpha activity along with decreases in delta power (Reference Hall, Thayer and Germain27). However, the cross-sectional correlation between DSR and alpha power that we observed is unable to prove whether hyperarousal processes could have been causally impairing delta power or vice versa.
Nonetheless, there are known differences in the hyperarousal observed in patients diagnosed with insomnia versus those with MDD (Reference Staner, Cornette and Maurice62). People with simple insomnia may have a primary problem of hyperarousal without evidence for a primary dysfunctional homeostatic drive (i.e. a low DSR). In addition, insomnia tends to also be associated with increases in beta power as well (Reference Hall, Thayer and Germain27,Reference Merica, Blois and Gaillard63). We found no evidence for beta or theta power on depression risk, nor did we find an affect of sleep efficiency or waking after sleep onset (by either diary self-report or PSG). It is therefore unlikely that overt sleep fragmentation with awakening affected depression risk. Our findings appear to be more specific to the 8–16 Hz range during NREM sleep; and do not support the possibility that at-risk people with poor sleep quality simply had a primary insomnia.
Moreover, we observed that subjects with both alpha power <6.5% (below the median) as well as DSR >1.4 (above the median) were resilient to any changes in depression scores during IFN-α therapy, indicating that both aspects of sleep are important for protection from depression risk. When either DSR was below the median (<1.4) or alpha power was above the median (>6.5%), then BDI-II increased during IFN-α therapy. Related to this, an increase in delta power with a reduction in both alpha and sigma is good predictor of antidepressant response (Reference Luthringer, Minot, Toussaint, Calvi-Gries, Schaltenbrand and Macher64). Thus, it may be the combination of both hyperarousal and impaired homeostatic sleep drive that could lead to depression vulnerability.
Notably, variation in sleep architecture, including delta power, appears to have a very strong genetic influence (Reference Linkowski65,Reference Ambrosius, Lietzenmaier and Wehrle66). We observed very strongly correlations between nights for DSR and for quantitative EEG power spectra, consistent with prior studies (Reference Buckelmuller, Landolt, Stassen and Achermann67,Reference Tucker, Dinges and Van Dongen68), indicating that these power spectra may be reliable and stable markers for individuals. Thus, these may be robust physiologic markers with utility in assessing depression vulnerability.
Normal NREM and delta sleep may result in restorative properties that decrease depression risk (Reference Datta and Maclean69). NREM disruptions can adversely influence oxidative stress (Reference Silva, Abilio and Takatsu70), cell proliferation (Reference Hairston, Little and Scanlon71,Reference Roman, Van der Borght and Leemburg72), excitatory/inhibitory balance (Reference Corner, Baker and van Pelt73,Reference Krueger and Obal74), hippocampal plasticity (Reference Guzman-Marin, Ying and Suntsova75), cortical synaptic plasticity (Reference Romcy-Pereira, Leite and Garcia-Cairasco76), and hypothalamic–pituitary–adrenal axis function (Reference Leproult, Copinschi and Buxton77,Reference Spath-Schwalbe, Gofferje, Kern, Born and Fehm78). Each of these could feasibly increase depression risk. Decreased delta power and increases in higher frequency power, which can be associated with elevated NREM metabolic activity in depression-related areas such as the reticular formation, the anterior cingulate cortex, and the orbitofrontal cortex (Reference Hofle, Paus, Reutens, Fiset, Gotman and Evans79). It is thus plausible that the low DSR indicates that delta sleep was insufficiently ‘restorative’ in the first NREM period, thereby influencing depression risk.
There are several ways in which poor sleep could vitiate the effects of IFN-α, although any mechanistic inferences are limited by the fact that we did not examine the physiology by which IFN-α induces depression. But because IFN-α can decrease BDNF levels (Reference Lotrich, Albusaysi and Ferrell80), it is feasible that insomnia-induced deficits in cell proliferation (Reference Hairston, Little and Scanlon71,Reference Roman, Van der Borght and Leemburg72) and plasticity (Reference Guzman-Marin, Ying and Suntsova75,Reference Romcy-Pereira, Leite and Garcia-Cairasco76) could have exacerbated the effect of inflammatory cytokines on neurogenesis. Also, IFN-α decreases hippocampal cell proliferation via elevated IL-1β levels (Reference Kaneko, Kudo and Mabuchi81); and social isolation likewise decreases central BDNF and neurogenesis – mediated in part by the inflammatory cytokines like IL-1β (Reference Ben Menachem-Zidon, Goshen and Kreisel82–Reference Koo and Duman84). Thus, both stress and inflammation share similar effects on growth factor function (Reference Peng, Chiou, Chen, Chou, Ky and Cheng85); and BDNF is required for the neuroprotective effects of antidepressants against lipopolysaccharide-induced apoptosis (Reference Peng, Chiou, Chen, Chou, Ky and Cheng85). Therefore, it makes sense that a pre-existing deficit in plasticity could be one potential mechanism by which poor sleep might exacerbate inflammatory affects on BDNF. Another possibility is that poor sleep affects the hypothalamic–pituitary–adrenal axis (Reference Leproult, Copinschi and Buxton77,Reference Spath-Schwalbe, Gofferje, Kern, Born and Fehm78), which is likely to aggravate the response to IFN-α (Reference Raison, Borisov, Woolwine, Massung, Vogt and Miller86). Insufficient delta power also influences the excitatory/inhibitory balance (Reference Corner, Baker and van Pelt73,Reference Krueger and Obal74), which may potentially exacerbate IFN-α effects on serotonin (Reference Raison, Borisov, Majer, Drake, Pagnoni and Miller87), dopamine (Reference Felger, Alagbe and Hu88), and glutamate (Reference Haroon, Woolwine and Chen89) systems. Whether some or all of these physiological interactions are truly involved remains to be determined. Moreover, further exacerbating any pre-existing sleep problems, inflammatory cytokines such as IFN-α can additionally decrease sleep efficiency, sleep continuity, and the total amount of stages 3 and 4 sleep (Reference Raison, Rye and Woolwine90). And of course, a variety of other cytokines can further influence sleep quality. Ultimately, a bi-directional relationship between sleep and inflammation in influencing depression-related physiology is highly plausible. Regardless, the mechanisms underlying these interactions remain speculative.
Despite advances in delineating the pathophysiology of inflammation-related depression (Reference Felger and Lotrich18), a critical clinical question that remains is how to best remediate vulnerability and prevent depression in the minority who are not yet resilient. Our findings indicate that low DSR and high alpha power may be a good physiological target for treatment as these elements of sleep are consistently present in MDD (Reference Armitage, Hoffmann, Trivedi and Rush13,Reference Kupfer, Frank and Ehlers15) and are worsened by stress and glucocorticoids (Reference Antonijevic and Steiger91).
Congruent with this conclusion, patients who already have higher DSR tend to do better with psychotherapies such as cognitive behavioural therapy (Reference Thase, Fasiczka, Berman, Simons and Reynolds92) or sleep deprivation (Reference Nissen, Feige, Konig, Voderholzer, Berger and Riemann93). Conversely, a low DSR in non-depressed patients is a robust predictor of depression relapse after successful psychotherapy (Reference Kupfer, Frank, McEachran and Grochocinski16,Reference Spanier, Frank, McEachran, Grochocinski and Kupfer30,Reference Buysse, Frank, Lowe, Cherry and Kupfer31), during maintenance psychotherapy (Reference Kupfer, Frank, McEachran, Grochocinski and Ehlers94), or during maintenance antidepressant treatment (Reference Reynolds, Buysse and Brunner32). DSR also tends to be a fairly robust trait (Reference Buysse, Hall and Tu12) that does not change over time even with some effective depression psychotherapies (Reference Buysse, Frank, Lowe, Cherry and Kupfer31,Reference Thase, Fasiczka, Berman, Simons and Reynolds92). Although some of these antidepressants interventions may not influence DSR, there is some evidence that insomnia-specific therapies such as cognitive behavioural therapy for insomnia could both improve DSR and decrease alpha power (Reference Krystal and Edinger39,Reference Cervena, Dauvilliers and Espa95). Thus, our results indicate that these types of specific insomnia therapies could be one avenue for preventing depression.
Another avenue for improving sleep is antidepressant medications. Different medications have different effects on various sleep parameters (Reference Wichniak, Wierzbicka and Jernajczyk17,Reference Sonntag, Rothe, Guldner, Yassouridis, Holsboer and Steiger96–Reference Argyropoulos and Wilson99). Some SSRIs and tricyclics can improve delta sleep (Reference Reynolds, Buysse and Brunner32,Reference Jindal, Friedman, Berman, Fasiczka, Howland and Thase100–Reference Pasternak, Reynolds and Houck103). In fact, SSRIs have been observed to improve DSR in both remitters and non-remitters (Reference Argyropoulos, Hicks and Nash104). The first SSRI observed to prevent depression in patients started on IFN-α was paroxetine (Reference Musselman, Lawson and Gumnick105), and paroxetine also decreases alpha power but increases DSR in non-depressed insomniacs (Reference Nowell, Reynolds, Buysse, Dew and Kupfer106), a plausible mechanism by which paroxetine could improve resilience. Conversely, some SSRIs can exacerbate increased alpha power (Reference Wichniak, Wierzbicka and Jernajczyk17). It will therefore be clearly important to assess sleep parameters if antidepressants are examined for depression prevention efficacy. Other potential depression prevention options include agomelatine, which improves DSR (Reference Salva, Vanier and Laredo107,Reference Quera-Salva, Lemoine and Guilleminault108), as well as ghrelin, which can decrease alpha power and improve the amount of slow wave NREM sleep (Reference Kluge, Gazea and Schussler109).
These speculations regarding medications to prevent depression are testable hypotheses. Our findings would specifically predict that instituting a sleep therapy that improves DSR and decreases alpha power before an inflammatory challenge (such as IFN-α therapy) would prevent depression. This treatment could be specifically targeted to people who are currently sleeping poorly (as defined by a low DSR and elevated alpha power assessed using qEEG). The model would likewise predict that any sleep therapies that worsen DSR and increase alpha power would not prevent depression (even if the intervention improves other sleep parameters).
However, it is important to note that our observations, although prospective, do not necessarily imply causality. Improving DSR and decreasing alpha power may not necessarily improve resilience, particularly if these sleep variables are simply manifestations of a more fundamental neurologic function. A clinical treatment trial would be needed to test this hypothesis. Another caveat to our findings is the need for replication, particularly given the limited sample size. In addition, to focus on the role of sleep, we examined a fairly resilient population screened to not have current psychiatric or substance abuse problems despite having HCV. It remains to be determined whether the findings generalise to more vulnerable HCV populations, to other types of inflammatory cytokine-associated depression (Reference Lotrich110), and/or to depression more broadly.
Nonetheless, it has been years since the initial observations that poor sleep quality antedates depression – and the suggestion that treating sleep could prevent depression (Reference Ford and Kamerow111). Determining potential specific targets for treatment has been the next step. Our results would predict that benzodiazepines, which may improve sleep latency and subjective sleep quality, but can decrease delta power and increase higher frequency power (Reference Bastien, LeBlanc, Carrier and Morin40,Reference Feinberg, Maloney and Campbell41), would be unlikely to prevent depression. Rather, using this specific medical population – patients treated with IFN-α – we find that subjects with a high DSR and lower alpha power are resilient towards developing inflammatory cytokine-associated depression. It will now be critical to determine if normalising these specific sleep parameters can result in depression resilience for other patients.
Acknowledgements
Both Drs. Lotrich and Germain contributed to the conception and design of this study, to the acquisition of data, to the analysis and interpretation of data, to the drafting of the article, and to the final approval of the manuscript submitted.
Financial Support
This study was supported by the National Institutes of Health (MH-R01-090250, UL1-RR-024153, UL1-TR-000005).
Conflicts of Interest
None.
Ethical Standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.