


Jervell and Lange-Nielsen syndrome,Reference Jervell and Lange-Nielsen 1 an inherited, arrhythmogenic, genetically heterogeneous,Reference Schulze-Bahr, Haverkamp and Wedekind 2 loss-of-function channelopathy, is the most severe variant of congenital long-QTc syndrome.Reference Schwartz, Spazzolini and Crotti 3 , Reference Goldenberg, Moss and Zareba 4 The mortality rate exceeds 50% among untreated affected children, and most die before the age of 15 years.Reference Tranebjaerg, Samson and Green 5 Equally disquieting, none of the current therapies offers a long-term absolute protection from sudden cardiac death regardless of the genotype or disease substrate. Importantly, sudden death may be a sentinel event.Reference Tranebjaerg, Samson and Green 5 – Reference Roden, Lazzara and Rosen 8
Clinically, long-QTc interval in a child with high-tone sensorineural deafness with some preservation of low tone from birth and recurrent syncopal episodes form the diagnostic hallmarks of the Jervell and Lange-Nielsen syndrome.Reference Jervell and Lange-Nielsen 1 – Reference Tranebjaerg, Samson and Green 5 These patients are at high risk for sudden death due to a distinctive form of ventricular tachyarrhythmia, Torsades de Pointes,Reference Tranebjaerg, Samson and Green 5 , Reference Dessertenne 9 which is often self-limited and is associated with syncopal episodes. It is important to note that such episodes may be clinically misinterpreted as a seizure disorder – epilepsy – but in fact relate to self-limited episodes of Torsades de Pointes that may degenerate into ventricular fibrillation and sudden death. This makes an accurate and a timely diagnosis of the syndrome imperative.
There is a proclivity for patients with Jervell and Lange-Nielsen syndrome to develop potentially lethal ventricular tachyarrhythmia in response to a sudden outpouring of catecholamine during periods of emotions or exercise.Reference Schwartz, Spazzolini and Crotti 3 , Reference Tranebjaerg, Samson and Green 5 , Reference Roden, Lazzara and Rosen 8 , Reference Kleinman, Glickstein and Shaw 10 , Reference Tranebjaerg, Bathen, Tyson and Bitner-Glindzicz 11 Traditionally, β-blocker and/or left cardiac sympathetic denervation are the mainstay therapies for patients with this syndrome.Reference Schwartz, Spazzolini and Crotti 3 , Reference Tranebjaerg, Samson and Green 5 , Reference Schwartz 6 Implantation of a cardiac defibrillator may provide an effective therapeutic alternative in some patients who continue to experience syncopal episodes despite β-blocker therapy and/or left cardiac sympathetic denervation or in those patients who experience sudden cardiac arrest.Reference Goldenberg, Moss and Zareba 4 , Reference Goel, Berger, Pelech and Dhala 12 , Reference Richter and Brugada 13
Genetic analysis in recent years has defined the molecular basis of Jervell and Lange-Nielsen syndrome. To date, mutation in two genes, KCNQ1 and KCNE1, have been identified to cause the syndrome.Reference Schulze-Bahr, Haverkamp and Wedekind 2 , Reference Tranebjaerg, Samson and Green 5 , Reference Neyroud, Tesson and Denjoy 14 , Reference Tyson, Tranebjaerg and Bellman 15 Both genes express in the heart and in the inner ear. Disease-causing gene mutation reduces the outward flowing, slowly activating component of the delayed rectifier repolarising current Iks.Reference Schulze-Bahr, Haverkamp and Wedekind 2 , Reference Tranebjaerg, Samson and Green 5 , Reference Neyroud, Tesson and Denjoy 14 , Reference Tyson, Tranebjaerg and Bellman 15 The latter plays a critical role to ensure a timely, orderly, effective, and rapid ventricular repolarisation,Reference Tranebjaerg, Samson and Green 5 , Reference Roden, Lazzara and Rosen 8 , Reference Roden and Spooner 16 , Reference Huang, Bitner-Glindzicz, Tranebjaerg and Tinker 17 as well as normal hearing.Reference Tranebjaerg, Samson and Green 5 , Reference Wangemann 18 , Reference Zdebik, Wangemann and Jentsch 19
This article reports the autosomal dominant mode of inheritance for both long-QT interval corrected for heart rate – long QTc – and for high-tone sensorineural deafness with some preservation of low tone in two successive generations from a highly inbred family with the Jervell and Lange-Nielsen syndrome; sequential electrocardiographic alterations before, during, and following spontaneous termination of the syncopal episode and the electrocardiographic changes that initiate Torsades de Pointes; unilateral high-tone sensorineural deafness that involves specifically the left ear in the father of the proband; long-term efficacy of β-blocker therapy alone in the severely symptomatic proband; and the natural history of the Jervell and Lange-Nielsen syndrome in this kindred with a different ethnic background based on a follow-up for 16 years in 59 living family members.
Methods
A total of 66 blood-related family members from three generations of a highly inbred family originating from Pakistan were tracked. The paternal and maternal grandparents of the proband and three first cousins who died before the proband was seen by us were evaluated on the basis of a detailed history alone.
A 12-lead scalar electrocardiogram was obtained from 59 living members from second and third generations including the proband. Hearing was tested by evoked response audiometry and a detailed clinical examination was performed for the proband, her parents, and two siblings – nuclear family. Clinical examination and audiometry were not performed for the 54 family members who were living in Pakistan – extended family.
A detailed history of symptoms such as seizure-like activity, syncopal episodes, or aborted cardiac arrest was obtained for each of the 59 living members. Age at onset, frequency of occurrence, duration of symptoms, and possible triggers such as emotional stress, exercise, swimming, or automobile driving were noted.Reference Schwartz, Spazzolini and Crotti 3 – Reference Tranebjaerg, Samson and Green 5 , Reference Tranebjaerg, Bathen, Tyson and Bitner-Glindzicz 11 Enquiries were made about consanguinity, clinically apparent hearing impairment, a history of otitis media, meningitis, skull trauma, use of ototoxic drugs such as streptomycin or quinine, and exposure to loud noise.
Measurements of QTc
QT interval was measured independently by two blinded investigators from five cardiac cycles as recommended for children and for adults.Reference Tranebjaerg, Samson and Green 5 , Reference Garson, Dick and Fournier 7 , Reference Goldenberg, Moss and Zareba 20 , Reference Priori, Napolitano and Schwartz 21 Heart rate-corrected QT intervals (QTc) were derived using the Bazett formula and mean QTc values were determined. The final values were averaged between the two observers.
Phenotypic characterisation
The following QTc intervals were used to identify the cardiac phenotype – affected – of the children: 470 and 480 ms or longer for boys and girls, respectivelyReference Tranebjaerg, Samson and Green 5 , Reference Garson, Dick and Fournier 7 ; and symptomatic adults: 440 and 460 ms or longer for men and women, respectively, associated with one or more of the following: adrenergic-triggered syncopal episode(s), documented Torsades de Pointes, T-wave alternans, and a positive family history of aborted cardiac arrest or sudden cardiac death below 35 years of age.Reference Tranebjaerg, Samson and Green 5 , Reference Tranebjaerg, Bathen, Tyson and Bitner-Glindzicz 11 , Reference Goldenberg, Moss and Zareba 20 , Reference Priori, Napolitano and Schwartz 21 Asymptomatic individuals with normal QTc – males <410 ms and females <420 ms – were considered as unaffected.Reference Tranebjaerg, Samson and Green 5 , Reference Priori, Napolitano and Schwartz 21 Electrocardiographic evidence of long-QTc interval, T-wave alternans, and Torsades de Pointes; audiometric evidence of high-tone sensorineural deafness with some preservation of low-tone hearing; and occurrence of adrenergic-triggered syncopal episode(s) formed the diagnostic criteria for Jervell and Lange-Nielsen syndrome.Reference Jervell and Lange-Nielsen 1 – Reference Tranebjaerg, Samson and Green 5 The criteria for phenotypic ascertainment were not age dependent.
Diagnostic scoring system
The proband and each member of the nuclear family were also evaluated by the scoring system proposed by Schwartz et al, where a score of 4 or more indicated a high probability, 2–3 an intermediate probability, and ≤1 a low probability for hereditary long-QT syndrome.Reference Schwartz, Moss, Vincent and Crampton 22
Follow-up
A total of 59 living members were followed up for 16 years, initially at 6-month intervals and later yearly. A detailed history, physical examination, and electrocardiogram were obtained at each visit for the proband and for each member of the nuclear family. In addition, 24-hour Holter monitoring was recorded and analysed for the proband. The parents of the proband provided the follow-up information for the 54 living members from the extended family who were living in Pakistan. A three-generation family tree was constructed (Fig 1).

Figure 1
I, II, III = First, second and third generation respectively. Each numeral represents a family member in each generation; PGP = Paternal grandparents; MGP = Maternal grandparents;
 $$$$
= Male and female, respectively;
$$$$
= Male and female, respectively;
 $$$$
= Proband;
$$$$
= Proband;
 $$$$
= Deceased grandparents. Clinical examination, electrocardiogram and audiometry were not done. There was no history of long-QTc-related cardiac events or deafness;
$$$$
= Deceased grandparents. Clinical examination, electrocardiogram and audiometry were not done. There was no history of long-QTc-related cardiac events or deafness;
 $$$$
= First cousins of the proband who had congenital deafness and died suddenly following adrenergic-triggered activities. Clinical examination, electrocardiogram and audiometry were not done. Historically, each had a severe phenotype of Jervell and Lange-Nielsen syndrome;
$$$$
= First cousins of the proband who had congenital deafness and died suddenly following adrenergic-triggered activities. Clinical examination, electrocardiogram and audiometry were not done. Historically, each had a severe phenotype of Jervell and Lange-Nielsen syndrome;
 $$$$
= Father of the proband. Electrocardiogram: mild-QTc prolongation. Audiometry: high-tone, sensori neural hearing loss involving specifically the left ear. There is no history of long-QTc-related cardiac events (mild Jervell and Lange-Nielsen syndrome phenotype);
$$$$
= Father of the proband. Electrocardiogram: mild-QTc prolongation. Audiometry: high-tone, sensori neural hearing loss involving specifically the left ear. There is no history of long-QTc-related cardiac events (mild Jervell and Lange-Nielsen syndrome phenotype);
 $$$$
= Affected family members living in Pakistan. Electrocardiogram: long-QTc. Audiometry and clinical examination were not done;
$$$$
= Affected family members living in Pakistan. Electrocardiogram: long-QTc. Audiometry and clinical examination were not done;
 $$$$
= Unaffected mother and one unaffected sibling of the proband. Electrocardiogram: normal-QTc Audiometry: normal hearing;
$$$$
= Unaffected mother and one unaffected sibling of the proband. Electrocardiogram: normal-QTc Audiometry: normal hearing;
 $$$$
= Affected sibling of the proband. Electrocardiogram: long-QTc. Audiometry: normal hearing;
$$$$
= Affected sibling of the proband. Electrocardiogram: long-QTc. Audiometry: normal hearing;
 $$$$
= Uncles, aunts, their spouses and first cousins of the proband. Electrocardiogram: normal-QTc. Audiometry: not done;
$$$$
= Uncles, aunts, their spouses and first cousins of the proband. Electrocardiogram: normal-QTc. Audiometry: not done;
 $$$$
= Consanguineous marriage.
$$$$
= Consanguineous marriage.
 $$$$
= Non-consanguineous marriage;
$$$$
= Non-consanguineous marriage;
 $$$$
= Twins (Zygosity not known).
$$$$
= Twins (Zygosity not known).
Results
The proband, born of a consanguineous marriage of first cousins (Fig 1), had her first syncopal episode at the age of 8 months following a bout of excessive crying. The frequency of adrenergic-triggered syncopal episodes increased gradually to five to six per day. These episodes were diagnosed initially as epilepsy. Anticonvulsant therapy had no effect.
The proband presented to us at the age of 4 years. During the initial visit, she was very apprehensive, became extremely agitated, and had a syncopal episode while an electrocardiogram was being recorded. A continuous recoding documented the sequential electrocardiographic changes that preceded the syncopal episode, occurred during syncope, and those that occurred after the end of the syncopal episode and were associated with spontaneous termination of Torsades de Pointes (Fig 2a). Another recording that was obtained sometime later, before β-blocker therapy was begun, highlighted the electrocardiographic changes that initiated Torsades de Pointes in the proband (Fig 2b). Complete blood count and peripheral blood smear examination showed microcytic hypochromic anaemia. Chest roentgenogram, urine analysis, serum electrolytes, transthoracic echocardiogram, and Doppler studies were all normal. Evoked response audiometry documented bilateral high-tone sensorineural deafness with some preservation of low-tone hearing.
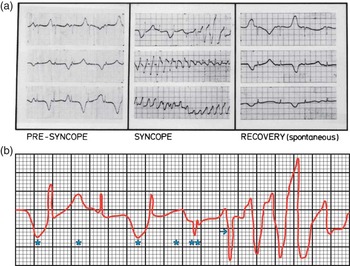
Figure 2 ( a ) Sequential electrocardiographic alterations before, during, and following: syncopal episode associated with Torsades de Pointes. Note: (i) Long-QTc interval, T-wave alternans, and bradycardia that precede syncopal episode; (ii) Torsades de Pointes associated with syncope; and (iii) Spontaneous termination of the syncopal episode and associated Torsades de Pointes. ( b ) Initiation of Torsades de Pointes. Note: Bradycardia; T-wave alternans (*) that precede an ectopic beat (**). The ectopic beat is followed by a morphologically abnormal ventricular complex (→) that initiates Torsades de Pointes.
Clinically, a diagnosis of Jervell and Lange-Nielsen syndrome was made and β-blocker therapy was begun – propranolol, 10 mg per oral route once every 6 hours. Following antiadrenergic therapy, the proband remained free of cardiac events, was discharged from the hospital 1 week later, and was followed up at regular intervals. Propranolol was later changed to Nadolol – 40 mg per oral route once daily.
Family pedigree of the proband (Fig 1; Table 1)
The nuclear family
The father of the proband had a QTc of 480 ms. Evoked response audiometry documented high-tone sensorineural deafness with some preservation of low tone. The deafness was unilateral and involved specifically the left ear. There was no history of clinically apparent hearing loss, syncopal episode, long-QTc-related cardiac events, rubella syndrome, skull trauma, use of ototoxic drugs, or exposure to loud noise. One asymptomatic sibling of the proband had long QTc and normal hearing. The mother and another sibling were asymptomatic with normal QTc and normal hearing.
Table 1 Clinical and electrocardiographic features of 27 affected members from two successive generations of a highly inbred family with Jervell and Lange-Nielsen syndrome.

ASYM = asymptomatic; ATS = adrenergic-triggered syncope; EKG = electrocardiogram; F = female; HR = heart rate; JLNS = Jervell and Lange-Nielsen syndrome; LQTc = long QTc interval; M = male; ms = millisecond; n/a = not available – patients died before the proband was seen; QTc = corrected QT interval; SND = sensorineural deafness; TDP = Torsades de Pointes
Diagnostic scoreReference Schwartz, Moss, Vincent and Crampton 22 : proband: 8; father of the proband: 4.5; affected sibling of the proband: 4; non-affected sibling of the proband and mother: each, 1
*Proband
**Father
***Sibling
****Hearing Tested
*****Hearing not tested
******Family history of sudden death below 30 years of age
The extended family
In all, seven family members died before the proband was seen. Both paternal and maternal grandparents died at old age from natural causes. None had a history of clinically apparent hearing impairment, syncopal episode, or sudden cardiac arrest. In addition, three first cousins of the proband with a history of deafness from birth and recurrent syncopal episodes died suddenly at young age following adrenergic-triggered activities (Table 1).
In all, 54 family members of the proband were alive and were living in Pakistan. There was a high prevalence of consanguinity in this kindred. Of the 10 parents in the second generation, seven were married to a first cousin (Fig 1, Table 1). It is interesting to note that 20 out of 29 offsprings were affected in six families where at least one parent was affected – consanguineous marriage: four; non-consanguineous marriage: two. By contrast, none of the 13 offsprings were affected in four families – consanguineous marriage: three; non-consanguineous marriage: one – where both parents were unaffected (Fig 1; Table 2).
Table 2 Affected parent(s) with or without consanguineous marriage: effect on phenotype in offspring.

C = consanguineous marriage; FC = first cousin; JLNS = Jervell and Lange-Nielsen Syndrome – LQTc as well as SND; LQTc = long QTc; NP = non-penetrant phenotype – QTc: normal, asymptomatic with no clinically apparent hearing loss; UA = unaffected – QTc: normal, asymptomatic and normal hearing; UR = unrelated
Affected: LQTc with or without SND
*Three first cousins of the proband: historically, severe Jervell and Lange-Nielsen syndrome phenotype. Each died suddenly following adrenergic-triggered activities
**Symptomatic proband with severe Jervell and Lange-Nielsen syndrome phenotype. Alive for 16 years with β-blocker therapy alone
Electrocardiographic alterations during syncopal episode (Figs 2a and b).
Diagnostic score (Table 1 footnote)
Follow-up: The severely symptomatic proband remained free of syncopal episodes and long-QTc-related cardiac events throughout the 16-year follow-up period while she maintained full compliance with β-blocker therapy. The last electrocardiogram at the end of the 16-year follow-up showed a resting heart rate of 58 beats/min and QTc of 580 ms. There were no ventricular ectopic beats or T-wave alternans. All 58 family members of the proband – including her father with a mild Jervell and Lange-Nielsen syndrome phenotype, her 22 affected but asymptomatic family members with only long QTc, which included her affected sibling, and 35 other asymptomatic family members with normal QTc interval – unaffected – remained asymptomatic throughout the follow-up period. None received antiadrenergic and/or antiarrhythmic therapy.
Discussion
Inheritance
Jervell and Lange-Nielsen syndrome is a hereditary disease. Since the original report by Fraser et al,Reference Fraser 23 , Reference Fraser, Froggatt and Murphy 24 it is commonly accepted that Jervell and Lange-Nielsen syndrome is inherited as an autosomal recessive trait.Reference Schulze-Bahr, Haverkamp and Wedekind 2 – Reference Roden, Lazzara and Rosen 8 , Reference Tranebjaerg, Bathen, Tyson and Bitner-Glindzicz 11 , Reference Neyroud, Tesson and Denjoy 14 – Reference Huang, Bitner-Glindzicz, Tranebjaerg and Tinker 17 However, James postulated that long QTc, the cardiac phenotype, could be inherited as autosomal dominant in some Jervell and Lange-Nielsen syndrome families.Reference James 25 This contention was confirmed in a genotyped family by Splawski et al, who reported an autosomal dominant inheritance for long QTc, but an autosomal recessive inheritance for high-tone sensorineural deafness.Reference Splawski, Timothy, Vincent, Atkinson and Keating 26 Contrary to the prevalent views, Matthews et al reported an autosomal dominant mode of inheritance for both cardiac and auditory phenotypes in four successive generations of a Jervell and Lange-Nielsen syndrome family.Reference Mathews, Blount and Townsend 27 Since then, three other investigators have reported an autosomal dominant mode of inheritance for this syndrome.Reference van der Straaten and Bruins 28 – Reference Furlanello, Maccà and Dal Palù 30
A high prevalence of consanguinity among parents of the affected offsprings in this study could suggest a quasi-dominant or a recessive inheritance for Jervell and Lange-Nielsen syndrome in this highly inbred family. However, each affected offspring in the third generation has at least one affected parent (Fig 1). Transmission of long QTc in two successive generations with involvement of both sexes, as well as transmission of high-tone sensorineural deafness with some preservation of low tone and long QTc from father to daughter, suggests an autosomal dominant mode of inheritance for both the cardiac and auditory phenotype in this study. Further, Fraser et al reported that in a family with Jervell and Lange-Nielsen syndrome the value of the ratio between the affected surviving sibs and the total number of sibs other than the proband is expected to be 0.25 for an autosomal recessive mode of inheritance.Reference Fraser, Froggatt and Murphy 24 However, this value is well above 0.25 for the family of the proband in the present study. Thus, observations of this study support the contentionReference Mathews, Blount and Townsend 27 – Reference Furlanello, Maccà and Dal Palù 30 that Jervell and Lange-Nielsen syndrome could be inherited as autosomal dominant in some affected families.
Consanguinity
A high prevalence of consanguinity has been reported in families with Jervell and Lange-Nielsen syndrome.Reference Tranebjaerg, Samson and Green 5 , Reference Roden, Lazzara and Rosen 8 , Reference Fraser, Froggatt and Murphy 24 In the present study, seven out of 10 parents in the second generation were married to a first cousin (Fig 1; Table 2). It is interesting to note that 20 out of 29 offsprings were affected in six families – consanguineous: four and non-consanguineous marriage: two – where at least one parent was affected. By contrast, none of the 13 offsprings were affected in four families where both parents were unaffected – three consanguineous and one non-consanguineous marriage. These observations (Fig 1; Table 2) seem to suggest that the presence of an affected parent rather than a consanguineous or non-consangineous marriage between the parents, most likely, plays an important role in the phenotypic expression of the offsprings in this highly inbred family with Jervell and Lange-Nielsen syndrome. Other factors such as promoter polymorphism, low penetrance, modifier genes, genetic imprinting, environmental stress, and ethnicity may further influence the phenotypic expression in the offsprings.Reference Tranebjaerg, Samson and Green 5 , Reference Roden, Lazzara and Rosen 8 , Reference Roden and Spooner 16 , Reference Priori, Napolitano and Schwartz 21 , Reference Crotti, Lundquist and Insolia 31 – Reference Fugate, Moss and Jons 35
Phenotype
Jervell and Lange-Nielsen syndrome is an allelic disorder. The dose of the mutant allele determines the phenotype in the affected individual.Reference Schwartz, Spazzolini and Crotti 3 , Reference Tranebjaerg, Samson and Green 5 Homozygous or compound heterozygous patients inherit two mutant allele, one from each parent. Mutations in KCNQ1 or KCNE1 gene, which cause the syndrome, usually produce truncated proteins that lack pore regions and hence do not form functional potassium (K+) channels in cardiac myocytesReference Roden, Lazzara and Rosen 8 , Reference Neyroud, Tesson and Denjoy 14 , Reference Tyson, Tranebjaerg and Bellman 15 – Reference Huang, Bitner-Glindzicz, Tranebjaerg and Tinker 17 , Reference Mohammad-Panah, Demolombe and Neyroud 36 as well as in marginal cells of striae-vascularis and in vestibular dark cells in the inner ear.Reference Schulze-Bahr, Haverkamp and Wedekind 2 , Reference Neyroud, Tesson and Denjoy 14 , Reference Wangemann 18 , Reference Zdebik, Wangemann and Jentsch 19 Consequently, there is an absence of Iks current and this leads to the severe phenotype in the proband and in her three deceased first cousins in the present study.Reference Schulze-Bahr, Haverkamp and Wedekind 2 , Reference Tranebjaerg, Samson and Green 5 , Reference Tranebjaerg, Bathen, Tyson and Bitner-Glindzicz 11 , Reference Huang, Bitner-Glindzicz, Tranebjaerg and Tinker 17
By contrast, the parents of the proband who presumably are obligate heterozygous and other heterozygous carriers among family members carry only one mutant allele. The normal allele tends to rescue the adverse functional consequences of the mutant allele. This, most likely, explains the mild phenotype in the heterozygous parents of the proband and other affected heterozygous individuals in the family who are asymptomatic and have QTc intervals that are within normal limits or show a mild prolongation.Reference Schwartz, Spazzolini and Crotti 3 , Reference Tranebjaerg, Samson and Green 5 , Reference Roden, Lazzara and Rosen 8 , Reference Tranebjaerg, Bathen, Tyson and Bitner-Glindzicz 11 , Reference Huang, Bitner-Glindzicz, Tranebjaerg and Tinker 17 , Reference Crotti, Lundquist and Insolia 31
Initiation of Torsades de Pointes
Sudden death or cardiac arrest that result from ventricular fibrillation triggered by Torsades de Pointes is a major concern in patients with Jervell and Lange-Nielsen syndrome. Continuous electrocardiographic recording in the proband during a syncopal episode showed that a marked prolongation of QTc interval associated with bradycardia, T-wave alternans, and ectopic beat preceded adrenergic-triggered Torsades de Pointes (Fig 2a). In these patients, an absence of Iks current prolongs the action potential duration in cardiac myocytes at the cellular level and sets the stage for development of early after depolarisations with triggered activity.Reference Roden and Spooner 16 , Reference Crotti, Spazzolini and Schwartz 33 The latter appears as ectopic beat with a pause on the surface electrocardiogram and triggers a morphologically abnormal ventricular complex that, in turn, initiates Torsades de Pointes (Fig 2b).Reference Roden and Spooner 16 Owing to the fact that triggered activity tends to slow down after a limited burst, Torsades de Pointes is usually self-limited and terminates spontaneously (Fig 2a). However, in some patients, Torsades de Pointes may degenerate into ventricular fibrillation, cardiac arrest, and sudden death.Reference Schwartz, Spazzolini and Crotti 3 , Reference Tranebjaerg, Samson and Green 5 , Reference Roden, Lazzara and Rosen 8 , Reference Roden and Spooner 16
The current therapeutic options
β-Blockers and/or left cardiac sympathetic denervation are the mainstay therapies for patients with Jervell and Lange-Nielsen syndrome.Reference Schwartz, Spazzolini and Crotti 3 – Reference Schwartz 6 The combination of pacing and β-blockers may be effective in some patients in whom bradycardia and pauses may facilitate potentially lethal Torsades de Pointes.Reference Dorostokar, Elder, Belhassen and Scheinman 37 , Reference Cooper, Stephenson, Berul, Walsh and Epstein 38
In recent years, several studies have demonstrated that implanted cardiac defibrillators may provide an effective alternative in the management of children and adolescents with Jervell and Lange-Nielsen syndrome.Reference Schwartz, Spazzolini and Crotti 3 , Reference Tranebjaerg, Samson and Green 5 , Reference Schwartz 6 , Reference Goel, Berger, Pelech and Dhala 12 , Reference Richter and Brugada 13 , Reference Cooper, Stephenson, Berul, Walsh and Epstein 38 Goldenberg et alReference Goldenberg, Moss and Zareba 4 reported a mortality rate of 35% in patients who received β-blocker alone but no mortality in those who received implanted cardiac defibrillator.Reference Goldenberg, Moss and Zareba 4 However, a number of complications have been reported with the use of these devices, which include, among others, psychological sequelae, electrical storms, and suicide attempts.Reference Tranebjaerg, Samson and Green 5 , Reference Schwartz 6 , Reference Goel, Berger, Pelech and Dhala 12 , Reference Dorostokar, Elder, Belhassen and Scheinman 37 – Reference Stephenson and Berul 41 Thus, the quest continues for a therapeutic regime that would ensure a long-term effective protection from sudden cardiac arrest and/or sudden death in patients with Jervell and Lange-Nielsen syndrome.
Natural history of Jervell and Lange-Nielsen syndrome in the present kindred
Clinical heterogeneity is common among the affected members in Jervell and Lange-Nielsen syndrome families.Reference Schulze-Bahr, Haverkamp and Wedekind 2 , Reference Tranebjaerg, Samson and Green 5 , Reference Tranebjaerg, Bathen, Tyson and Bitner-Glindzicz 11 , Reference Huang, Bitner-Glindzicz, Tranebjaerg and Tinker 17 In the present report, the proband and her three first cousins had a severe phenotype. None of the three first cousins received antiadrenergic or antiarrhythmic therapy and each died suddenly following adrenergic-triggered activities. By contrast, the severely symptomatic proband who received only β-blocker and maintained full compliance with the therapy did not experience syncopal episodes, Torsades de Pointes, or sudden cardiac arrest throughout the 16-year follow-up period. Similarly, 58 other family members remained free of long-QTc-related cardiac events during the entire follow-up period without any form of antiadrenergic and/or antiarrhythmic therapy. Thus, observations of the present study support the contention that obligate heterozygous parents, and the majority of heterozygous affected members in families with Jervell and Lange-Nielsen syndrome, usually have a mild phenotype.Reference Schwartz, Spazzolini and Crotti 3 , Reference Tranebjaerg, Samson and Green 5 , Reference Roden, Lazzara and Rosen 8 , Reference Tranebjaerg, Bathen, Tyson and Bitner-Glindzicz 11 , Reference Huang, Bitner-Glindzicz, Tranebjaerg and Tinker 17 , Reference Mohammad-Panah, Demolombe and Neyroud 36
Unilateral deafness in families with Jervell and Lange-Nielsen syndrome
The father of the proband presented with high-tone sensorineural deafness with some preservation of low tone that was unilateral and involved specifically the left ear. This unusual clinical manifestation has been reported to date in three other families with Jervell and Lange-Nielsen syndrome.Reference Mathews, Blount and Townsend 27 , Reference van der Straaten and Bruins 28 , Reference Duggal, Vesely and Wattanasirichaigoon 42 Of particular interest is the genotyped family with KCNE1 mutation reported by Duggal et alReference Duggal, Vesely and Wattanasirichaigoon 42 where the heterozygous mother had long-QTc interval, syncope, and unilateral deafness that involved specifically the left ear. However, Duggal et alReference Duggal, Vesely and Wattanasirichaigoon 42 and othersReference Mathews, Blount and Townsend 27 , Reference van der Straaten and Bruins 28 mentioned the unilateral deafness only in passing. The underlying basis for unilateral deafness in families with Jervell and Lange-Nielsen syndrome that involves specifically the left ear remains thus far unexplained.
Speculation
Converging evidence from several studies suggests that sound perception and cochlear processing of the sound waves are asymmetrical between the left and right ear.Reference Sininger and Cone-Wesson 43 Histopathological studies of the ear from patients with Jervell and Lange-Nielsen syndrome, as well in knockout KCNQ1 and KCNE1 mice, demonstrate a distinct asymmetry of the degree of degenerative changes between the basal and the apical cochlear areas.Reference Friedmann, Fraser and Froggatt 44 – Reference Rivas and Francis 46
Unilateral high-tone sensorineural deafness that specifically involved the left ear only in father of the proband in the present study could suggest that, as compared with the right ear, the left ear perhaps is highly sensitive to the adverse effects of the disease-causing mutant gene and hence is unable to maintain the normal homeostasis of the otic endolymph quantitatively and qualitatively.Reference Sininger and Cone-Wesson 43 , Reference Rivas and Francis 46 , Reference Hone and Smith 47 Consequently, the perception and the processing of the sound waves by the left ear is impaired and leads to a partial, high-tone, sensorineural deafness. Other factors that influence the clinical expression of mutant gene such as low penetrance, modifier genes, promoter polymorphism, environmental stress, and ethnicityReference Tranebjaerg, Samson and Green 5 , Reference Roden, Lazzara and Rosen 8 , Reference Roden and Spooner 16 , Reference Priori, Napolitano and Schwartz 21 , Reference Crotti, Lundquist and Insolia 31 – Reference Fugate, Moss and Jons 35 may further compound the degree of hearing loss. Thus, an interplay of these complex intrinsic and extrinsic factors may explain the unilateral hearing loss that specifically involved the left ear in father of the proband in the present study, in the heterozygous mother of the severely affected proband reported by Duggal et al,Reference Duggal, Vesely and Wattanasirichaigoon 42 and by two other investigators in families with Jervell and Lange-Nielsen syndrome.Reference Mathews, Blount and Townsend 27 , Reference van der Straaten and Bruins 28
The study limitations
The present study lacks genetic testing in the proband and in the at-risk family members. That genetic testing would identify the affected gene and establish the disease-causing mutation, as well as establish the carrier status among at-risk family members, some of whom could be silent carriers, is now well recognised.Reference Tranebjaerg, Samson and Green 5 , Reference Roden and Spooner 16 , Reference Priori, Napolitano and Schwartz 21
Identification of silent carriers and the carrier status in the affected individuals among the at-risk family members is extremely important as drugsReference Tranebjaerg, Samson and Green 5 , Reference Roden, Lazzara and Rosen 8 and anaesthetic agents that block Ikr current,Reference Kies, Pabelick, Hurley, White and Ackerman 48 , Reference Adu-Gyamfi, Said, Choudhary, Abomelha and Sanyal 49 in the presence of an intrinsic decrease in Iks current, may further prolong ventricular repolarisation and compromise myocardial repolarisation reserveReference Roden 50 to a sufficient degree and lead to Torsades de Pointes, cardiac arrest, and sudden death. Equally important, carriers could transmit a potentially lethal disease to the offspring.
However, carrier testing for at-risk family members requires that the gene and the disease-causing mutation are first identified in the proband.Reference Tranebjaerg, Samson and Green 5 Accordingly, genetic testing was planned for the proband, her immediate family members, and those from the extended family living in Pakistan. Unfortunately, the parents refused genetic testing. Finally, although the present observational study does not allow a definitive answer, it strongly emphasises the need for further long-term studies that involve large, uniform, cohorts of Jervell and Lange-Nielsen syndrome with different ethnic backgrounds.
Conclusion
We conclude that both cardiac and auditory phenotypes are inherited as autosomal dominants in this highly inbred family with Jervell and Lange-Nielsen syndrome; marked clinical heterogeneity exists in the kindred; unilateral, high-tone sensorineural deafness with some preservation of low tone that involves specifically the left ear could manifest in some affected heterozygous individuals with the syndrome; occasionally, β-blocker alone could provide a long-term effective therapy in a severely symptomatic patient with Jervell and Lange-Nielsen syndrome; and a 16-year follow-up of the kindred supports the contention that a mild phenotype is seen in the majority of the affected family members.
Acknowledgement
The authors thank Dr Abdul Aziz Ashoor, King Fahd Teaching Hospital of the University at Al-Khobar, Kingdom of Saudi Arabia, for audiometry studies; Ms Sharon Levine (United States of America) and Dr Somnath Bhattacharya and Mr Kapil Kumar (India) for their help in the preparation of the manuscript. The authors wish to express special appreciation and gratitude to the late Professor Charles S. Kleinman, Director Fetal Cardiology, Columbia University Medical Center, New York (United States of America), for his time, efforts, and invaluable contribution towards this manuscript. The authors would like to dedicate this article to the late Professor Charles S. Kleinman.




