Introduction
The production of pharmaceutical proteins by transgenic animals is one of the major successes of biotechnology (Houdebine, Reference Houdebine2000, Reference Houdebine2009). The production of pharmaceutical proteins in the mammary gland may have applications similar to those in other tissues, blood, and urine because milk proteins are naturally secreted (Houdebine, Reference Houdebine2009). However, transgenic animals show various levels of transgene expression due to differences in chromosomal location as a result of the random integration of a transgene, copy number of a transgene, and use of inadequate promoters (Houdebine, Reference Houdebine2009).
To improve production efficiency of therapeutic proteins in milk, gene-targeting methods using homologous gene recombination techniques have been developed (McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000). In particular, the ovine α1 (I) procollagen locus was targeted with a targeting vector containing the IRES-neo gene and ovine β-lactoglobin-driven human α1-antitrypsin, which was located behind the IRES-neo cassette, for direct expression in the lactating mammary gland. Using this approach, the authors obtained high expression (650 μg/ml) of α1-antitrypsins in milk. They used the β-lactoglobin promoter with an exogenous promoter to express the α1-antitrypsin transgene (McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000). Recently, it has been shown, by means of a chromatin-immunoprecipitation-based microarray method, that distal enhancers are very important for transcriptional gene regulation and tissue-specific gene expression (Heintzman et al., Reference Heintzman, Hon, Hawkins, Kheradpour, Stark, Harp, Ye, Lee, Stuart, Ching, Ching, Antosiewicz-Bourget, Liu, Zhang, Green, Lobanenkov, Stewart, Thomson, Crawford, Kellis and Ren2009; Visel et al., Reference Visel, Blow, Li, Zhang, Akiyama, Holt, Plajzer-Frick, Shoukry, Wright, Chen, Afzal, Ren, Rubin and Pennacchio2009). It has also been reported that the polypeptide precursors of milk proteins contain a hydrophobic signal peptide at their N terminus, which is necessary for translocation across the endoplasmic reticulum (Mercier & Gaye, Reference Mercier and Gaye1980; Burgoyne & Duncan, Reference Burgoyne and Duncan1998). According to this previous report, an endogenous promoter including an enhancer and signal sequence of the transgene is necessary to express a transgene at the same level as an endogenous gene. Due to the insertion of an exogenous gene by gene targeting, especially by the knock-in method, it is possible to express an exogenous gene in a host organism using all of the gene-regulatory DNA sequences containing a promoter and enhancers of the genomic locus.
It has been reported that 9–11 g/l of β-casein protein are produced in skimmed milk in the bovine (Eigel et al., Reference Eigel, Butler, Ernstrom, Farrell, Harwalkar, Jenness and Whitney1984). However, β-casein knockout mice survive normally and could even become pregnant and nurse their young (Kumar et al., Reference Kumar, Clarke, Hooper, Horne, Law, Leaver, Springbett, Stevenson and Simons1994). These results suggested that when a specific transgene is inserted into the β-casein gene locus by knock-in, the transgene can be expressed at the same level as β-casein by a β-casein gene-regulatory sequence containing an endogenous promoter and enhancer. Also, human fibroblast growth factor 2 (FGF2) is involved in cell growth, migration, wound healing, and tissue repair (Bikfalvi et al., Reference Bikfalvi, Klein, Pintucci and Rifkin1997). These report indicated that FGF2 may serve as a potential therapeutic agent. However, FGF2 is not a secretary protein and is localized primarily in the cytoplasm (Bikfalvi et al., Reference Bikfalvi, Klein, Pintucci and Rifkin1997).
Gene targeting is a powerful technique that involves homologous recombination to change and precisely replace an endogenous gene locus with an exogenous gene (Houdebine, Reference Houdebine2000; Laible & Alonso-González, Reference Laible and Alonso-González2009). Gene-targeting technology has been widely used to identify gene function and to generate mouse disease models. Among domestic animals, the first gene-targeted sheep was generated by somatic cell nuclear transfer (McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000) because embryonic stem cells, which have been used to generate gene-targeted mice, were not available for domestic animals. Although the development of effective gene-targeting technology is very important for production of domestic transgenic animals for biomedical research, the efficiency of gene targeting in somatic cells is lower than that in mouse embryonic stem cells (Denning & Priddle, Reference Denning and Priddle2003; Laible & Alonso-González, Reference Laible and Alonso-González2009).
To date, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 systems have been developed for gene targeting (Porteus & Carroll, Reference Porteus and Carroll2005; Wei et al., Reference Wei, Liu, Yu, Zhang, Gao and Jiao2013). Knockout rats and mice have been generated by introducing non-homologous end-joining-mediated deletions or insertions at a target site using ZFNs, TALENs, and CRISPR/Cas9 systems (Geurts et al., Reference Geurts, Cost, Freyvert, Zeitler, Miller, Choi, Jenkins, Wood, Cui, Meng, Vincent, Lam, Michalkiewicz, Schilling, Foeckler, Kalloway, Weiler, Ménoret, Anegon, Davis, Zhang, Rebar, Gregory, Urnov, Jacob and Buelow2009; Cui et al., Reference Cui, Ji, Fisher, Wu, Briner and Weinstein2011; Sung et al., Reference Sung, Jin, Kim and Lee2014). Currently, engineered artificial endonucleases make it possible to easily implement homologous recombinants using donor DNA and gene knockout in domestic animals (Tan et al., Reference Tan, Carlson, Lancto, Garbe, Webster, Hackett and Fahrenkrug2013; Ni et al., Reference Ni, Qiao, Hu, Zhao, Regouski, Yang, Polejaeva and Chen2014). In addition, the CRISPR/Cas9 system has been used to generate a knockout pig (Hai et al., Reference Hai, Teng, Guo, Li and Zhou2014; Whitworth et al., Reference Whitworth, Lee, Benne, Beaton, Spate, Murphy, Samuel, Mao, O'Gorman, Walters, Murphy, Driver, Mileham, McLaren, Wells and Prather2014). Nevertheless, it has not been reported that transgenic cattle can produce pharmaceutical proteins by means of the CRISPR/Cas9 system.
The aim of this study was to develop a gene-targeting knock-in system for expression of the human FGF2 fusion gene in the bovine β-casein gene locus with the ultimate goal of creating transgenic animals so that exogenous gene expression is not affected by the position effect. This approach is expected to significantly improve the level of expression. Human FGF2 mRNA was expressed in mouse mammary epithelial cells (HC11 cells) transfected with the knock-in vector; the protein was secreted into the culture medium. Furthermore, we established gene-targeted bovine fibroblast cell lines with integration into the bovine β-casein gene locus by CRISPR/Cas9-mediated homologous recombination for somatic cell nuclear transfer (SCNT); thus, we generated bovine transgenic cloned embryos by SCNT. These results indicated that the knock-in system was successfully established to ensure stable expression of a transgene by the gene-regulatory sequences of the endogenous β-casein gene. This approach could be used to create transgenic cattle for production of therapeutic proteins in the mammary glands.
Materials and methods
Construction of the knock-in vector
Homologous arms were amplified by polymerase chain reaction (PCR) from dairy cattle genomic DNA. The 5.98-kb 5′-arm fragment of the bovine β-casein gene was cloned by amplification using a forward primer with an additional NotI site (CGGC- GGCCGCATGCTGAAGCTGAAACTCCT) and a reverse primer with an additional SalI site (GTCGA- CGAGTTCTTCCAGCTAAAGAG). The 2.08-kb 3′-arm fragment was amplified using a forward primer with an additional BamHI site (GGATCCATTGTG- GAAAGCCTTTCAAGCAGTGAG) and a reverse primer with an additional SalI site (GTCGACCT- CTGTTTGCTGCTGTTCCTCACTCTG). The PGKneo-polyA fragment for positive selection was isolated from the pKJ2 neo plasmid (Gibco BRL; Grand Island, NY, USA) with EcoRI and BamHI digestion. The human FGF2 cDNA was amplified from a human hepatocellular liver carcinoma cell line (HepG2, ATCC No. HB-8065) by reverse transcription (RT)-PCR and specific primers (forward primer with an additional KpnI site; GGTACCATGGCAGCCGGGAGCATC, rev- erse primer with an additional SmaI site; CCCGGG- AAATCAGCTCTTAGCAGAC). The bovine growth hormone polyA (BGH polyA) signal sequence was amplified by PCR using a forward primer with an additional SmaI site (CCCGGGCGACTGTGCCTTCT- AGTT) and a reverse primer with an additional EcoRI site (GAATTCCATAGAGCCCACCGCAT) from the pRc/CMV plasmid (Invitrogen; Carlsbad, CA, USA) as a template. Oligonucleotides encoding F2A (sense: CTCGAGAAAATTGTCGCTCCTGTCAAAC- AAACTCTTAACTTTGATTTACTCAAACTGGCTG- GGGATGTAGAAAGCAATCCAGGTCCAGGTACC, antisense: GGTACCTGGACCTGGATTGCTTTCTAC- ATCCCCAGCCAGTTTGAGTAAATCAAAGTTAA- GAGTTTGTTTGACAGGAGCGACAATTTTCTCG- AG) were synthesized (Bioneer; Daejeon, Korea) and annealed. The annealed F2A DNA was subcloned into the XhoI–KpnI sites of a pBSK+ plasmid. All the fragments for PCR amplification were subcloned into pGEM-T easy vector (Promega Co.; Madison, WI, USA) or pBluescript SK+ (pBSK+) plasmid (Promega), and the primary structure of all fragments was confirmed by sequencing. To construct the knock-in vector, first, F2A (XhoI–KpnI) and human FGF2 cDNA (KpnI–SmaI) fragment were ligated into the XhoI–SmaI site of the pBKS(+) plasmid via three-fragment ligation to produce the pBKS–F2A–hFGF2 plasmid. The 5′-arm fragment (NotI–SalI) was inserted into the NotI–XhoI site of the pBKS–F2A–hFGF2 plasmid to produce the pBKS–5′-arm–F2A–hFGF2 plasmid. Then the 5′-arm–F2A–hFGF2 fragment from the pBKS–5′-arm–F2A–hFGF2 plasmid was inserted into the NotI–SmaI site of pMCDT–A (A+T/pau) plasmid (Gibco BRL, Grand Island, NY, USA) by subcloning to produce the pMCDT–5′-arm–F2A–hFGF2 plasmid. The BGH polyA (SmaI–EcoRI) and PGKneo (EcoRI–BamHI) fragment were ligated into the SmaI–BamHI site of the pBKS(+) plasmid via three-fragment ligation to produce the pBKS–BGHp–PGKneo plasmid. Next, BGH polyA–PGKneo (SmaI–BamHI) from pBKS–BGHp–PGKneo plasmid and the 3′-arm (BamHI–SalI) fragment were ligated into the pBSK(+) plasmid via three-fragment ligation to produce the pBSK–BGHp–PGKneo–bβC3′-arm plasmid. Finally, to generate the knock-in vector, the BGHp–PGKneo–bβC3′-arm fragment was inserted into the SmaI–XhoI site of the pMCDT–5′-arm–F2A–hFGF2 plasmid (Fig. 1). These knock-in vectors were digested with the NotI restriction enzyme to linearize them prior to transfection.

Figure 1 The knock-in strategy in the bovine β-casein gene locus. The diagram shows homologous recombination of the knock-in vector in the bovine β-casein gene locus. The PGKneo gene and DT-A gene without the polyA signal sequence were used as positive and negative selection markers, respectively. The PCR primer pairs used for detecting homologous recombination events are shown by name.
Isolation and culture of bovine fibroblasts
Bovine fibroblast cells were obtained from ear tissues of 1-month-old dairy cattle according to a modified method for isolation of porcine ear fibroblasts (Ahn et al., Reference Ahn, Kim, Kim, Lee, Heo, Kang, Kang, Lee, Lee, Kim, Nho, Hwang, Woo, Park, Park and Shim2011). The fibroblasts were cultured in the growth medium (Dulbecco's modified Eagle medium [DMEM], 10% fetal bovine serum [FBS], 1× non-essential amino acids, 1× sodium pyruvate, 10–4 M β-mercaptoethanol, 100 units/ml penicillin, 100 μg/ml streptomycin) for maintenance and subculture. For storage, the fibroblasts were frozen in DMEM with 10% FBS and 10% dimethyl sulfoxide (DMSO).
Transfection of the knock-in vector into the fibroblasts
The transfection was conducted using electroporation as follows. The cultured cells were harvested by incubation with 0.25% trypsin and resuspended in Ham's F10 medium (HyClone Co.; Logan, UT, USA) at the density of 5 × 106 cells per 0.4 ml. The cell suspension (400 µl) was electroporated in a 4-mm cuvette using 10 µg of the linearized knock-in vector only or 5 µg of the linearized knock-in vector, 2.5 μg of sgRNA expression vector, and 2.5 μg of Cas9 nuclease expression vector mixture DNA with four 1-ms pluses using 450-V capacitive discharges on the BTX Electro-cell manipulator (ECM 2001, BTX; Holliston, MA, USA). After electroporation, the cuvette was placed on ice for 10 min. The cells in the cuvette were resuspended in 10 ml of the medium, which was dispensed in a 24-well plate (4000 cells/well), followed by further culture. After 48 h, selection was performed for 10–12 days using 500 µg/ml G418 (Gibco BRL; Grand Island, NY, USA). After selection, the stand-alone colonies were passaged into 6-well plates, and were further cultured for analysis of transgene-positive colonies.
Screening and PCR analysis of knock-in colonies
Knock-in colonies were identified by first and second PCRs. A 200-μl aliquot of a cell suspension from a 6-well plate was centrifuged at 800 g for 5 min at 4°C for the screening of G418-resistant colonies by the first PCR. The cells were resuspended in 100 μl of distilled water containing 0.05 mg/ml proteinase K (Roche, Indianapolis, IN, USA). In order to extract genomic DNA, the cells were incubated at 55°C for 130 min and heated to 98°C for 10 min to inactive proteinase K. The first PCR was performed in a 50 μl volume containing 20 μl genome extract, 0.1 M Neo sense (CGATCAGGATGATCTGGACGAAGAGCATCA) and FGF KI Sc antisense 3 (GCTGATAAATGATCAGATGAGGATTAGTGC) primers located outside the recombination region, 1× PCR buffer, 1.25 U Ex Taq DNA polymerase (TaKaRa Co., Tokyo, Japan), and 200 μM of each dNTP. PCR amplification was run for 37 cycles of 20 s at 95°C for denaturation, 40 s at 66°C for annealing, and 3 min at 72°C for extension. Positive colonies from the first PCR were subcultured in 10-cm dishes. The cells were then frozen in medium (culture medium containing 10% DMSO) for further analyses. Genomic DNA was isolated from the cell clones positive in the first PCR analysis by means of a GenElute Mammalian Genomic DNA Miniprep kit (Sigma-Aldrich, St. Louis, MO, USA) to accurately analyze the knock-in colonies by the second PCR. The second PCR amplification was conducted using 3′-arm, 5′-arm, and targeted or non-targeted PCR. The 3′-arm PCR was conducted under the same conditions as the first PCR. The 5′-arm PCR was performed with 50 ng of genomic DNA, bβc 5′-Sc sense 1 primer (ATTCTTCAAGTGATGGGAGTACCAGACCAC) and Neo 5′-2 antisense primer (TGCTAAAGCGCATGCTCCAGACTGCCTTGG), 1× PCR buffer, 1 U KOD FX Neo polymerase (Toyobo Co., Osaka, Japan), and 200 μM of dNTP mixture in a total volume of 50 μl. PCR amplification was performed using step-down cycles as follow: 2 min at 94°C (pre-denaturation), 5 cycles of 10 s at 98°C, 4 min at 74°C; 5 cycles of 10 s at 98°C, 4 min at 72°C; 5 cycles of 10 s at 98°C, 4 min at 70°C; 18 cycles of 10 s at 98°C, 4 min at 68°C; and final extension of 7 min at 68°C. To identify targeted and non-targeted alleles in the knock-in cells, PCR was conducted using 50 ng of genomic DNA isolated from G418-resistant colonies, 10 pmol each of pβE2 5′-sense 1 primer (GATCAAACCTGGGCTCCCCTGGAAGCA) and FGF KI Sc antisense 3 (GCTGATAAATGATCAGATGAGGATTAGTGC), 1× PCR buffer, 1 U KOD FX Neo polymerase (Toyobo Co.), and 200 µM of the dNTP mixture in a total volume of 50 μl. PCR amplification was performed using step-down cycles as follows: 2 min at 94°C (pre-denaturation); 5 cycles of 10 s at 98°C, 4 min at 74°C; 5 cycles of 10 s at 98°C, 4 min at 72°C; 5 cycles of 10 s at 98°C, 4 min at 70°C; 20 cycles of 10 s at 98°C, 4 min at 68°C; final extension of 7 min at 68°C. PCR products (20 μl) were confirmed by electrophoresis on a 0.8% agarose gel.
Southern blot analysis of knock-in colony
Final confirmation of knocked-in cells was performed by Southern blot analysis. Fifteen μg of the genomic DNA was digested with XbaI (TaKaRa Co.). The DNA was electrophoresed on 0.8% agarose gel, and was then transferred on nitrocellulose membrane (Bio-Rad Co., USA). The DNA was cross-linked onto the membrane by using an ultraviolet (UV) light crosslinker, followed by hybridization in the PerfectHyb™ plus hybridization buffer (Sigma Chemical Co., USA) at 68°C for 15 h. A probe was prepared by using a 509-bp DNA fragment containing the bovine β-casein exon 7, random primer DNA labelling kit (TaKaRa Co.) and (α-32P) dCTP (110 TBq/mmol, Amersham., England). A neo probe was also prepared by using a 600 bp/PstI DNA fragment from the pKJ2 plasmid. After hybridization, the membrane was washed four times with 0.3% SSC and 0.1% SDS (w/v) at 68°C for 20 min, exposed on X-ray film (Sigma Chemical Co., St Louis, MO, USA) at –80°C for 5 days, and then developed.
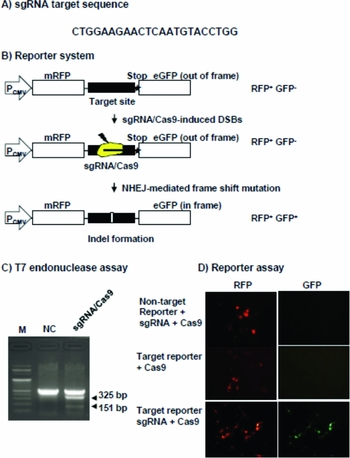
The CRISPR/Cas9 system and red fluorescent protein (RFP)–green fluorescent protein (GFP) reporter plasmid
Single guide RNA (sgRNA) expression vector (pRGEN_Bovine CSN2_U6_SG plasmid) harboring the U6 promoter designed to cleave the exon 3 region of the bovine β-casein gene was purchased from ToolGen Inc. (Seoul, Korea). The Cas9 nuclease expression vector with the CMV promoter was obtained from ToolGen Inc. (Seoul, Korea) (Cho et al., Reference Cho, Kim, Kim and Kim2013). The target and non-target RFP-GFP reporter plasmid (pRG2S_Bovine CSN_CMV plasmid) for an episomal reporter assay of CRISPR/Cas9 activity was purchased from ToolGen. The target RFP and GFP reporter plasmids were designed for expression of RFP and GFP fusion proteins by inserting sgRNA target sites (CTGGAAGAACTCAATGTACCTGGTT) between the RFP and GFP genes. The non-target RFP and GFP reporter plasmids had an sgRNA non-target sequence from the exon 7 of bovine β-casein gene.
The T7 endonuclease I assay for validation of the CRISPR/Cas9 system
The sgRNA target region was amplified from genomic DNA by PCR using gene-specific primers (sense, GCTGTGTCTATTCAACACAGGATGATACTC; antisense, AACACTATCTTACCTCACTGCTTGAAAGGC). The amplicons were purified using the Qiagen quick gel extraction kit (Qiagen, Hilden, German) denatured by heating and then annealed in hybridization buffer (10 mM Tris–HCl [pH 8.5], 75 mM KCl, 1.5 mM MgCl2) for formation of heteroduplex DNA. After treatment with T7 endonuclease I (10 U, New England BioLabs, Ipswich, MA, USA) at 37°C for 30 min, the resulting fragments were subjected to electrophoresis on a 2% agarose gel and visualized by staining with ethidium bromide.
CRISPR/Cas9 system-induced mutagenesis at an episomal target site
MAC-T cells were received from KonKuk University. Originally, the Mac-T cells were provided by F.Q. Zhao (University of Vermont, Burlington, USA). The cells were cotransfected with an sgRNA expression vector (1.25 µg), Cas9 nuclease expression vector (1.25 µg), and the target or non-target RFP-GFP reporter plasmid (2.5 µg) in a 6-cm culture dish using the jetPEI transfection reagent (Polyplus-transfection, Illkirch, France) according to the manufacturer's instructions. Thirty-six hours after transfection, RFP and GFP expression levels were examined under a fluorescence microscope (Nikon; Tokyo, Japan) and photographs were captured for analysis.
Cell culture of HC11 cells, transfection, and lactogenic hormone-induced differentiation
The HC11 cells were cultured in a growth medium: RPMI1640 (HyClone Co.; Logan, UT, USA) containing 10% FBS (HyClone Co.), 100 U/ml penicillin, 100 µg/ml streptomycin (HyClone Co.), 5 µg/ml insulin (Sigma-Aldrich), and 10 ng/ml epidermal growth factor (EGF; Invitrogen Co.). Transfection of the knock-in vector was performed as follows. The HC11 cells were seeded in 35-mm dishes at the density of 2.4 × 105 cells/dish, and the culture medium was replaced with 1.8 ml of fresh medium the next day. The cells were then transfected using the jetPEI transfection reagent (Polyplus-transfection) according to the manufacturer's instructions using 4 µg of the knock-in vector and pSV2neo as neo resistance gene for G418 selection. After 24 h, the cells were passaged into 10-cm culture dishes and selection was performed for 10 days using 300 µg/ml of G418 (Gibco BRL). After 10 days, colonies were picked using a colony cylinder and were proliferated in the culture medium. For lactogenic hormone induction, the HC11 cells (105 cells per six wells) as a negative control and HC11-hFGF2 cells were cultured in the growth medium. The lactogenic hormone induction of HC11 cells was described previously (Doppler et al., Reference Doppler, Groner and Ball1989). Briefly, the cells were grown to confluence and were maintained for 3 days in the growth medium. The medium was removed and the cells were washed three times with RPMI to remove the remaining serum. The cells were then cultured with induction medium [RPMI1640 (HyClone Co.) containing 100 U/ml penicillin, 100 µg/ml streptomycin, 5 µg/ml insulin (Sigma-Aldrich), 1 µM dexamethasone (DEX; Sigma), 5 µg/ml prolactin (Sigma)] for 3 days without changing the medium.
Isolation of total RNA and RT-PCR
Total RNA was isolated from HC11 cells transfected with the human FGF2 knock-in vector using the RNeasy mini kit (Qiagen, Hilden, Germany). The total RNA was then treated with DNase I, after which reverse transcription was performed at 42°C for 90 min using 5 µg of total RNA, random primers, and Superscript II RNase H-Reverse transcriptase (Invitrogen). Two microlitres of cDNA was utilized for PCR amplification of hFGF2 (sense primer, TCTGGCCCTTGCAAGAG; antisense primer, CACATTTAGAAGCCAGTAAT). The PCR reaction mixtures for hFGF2 contained 2.5 U Go Taq polymerase (Promega), 1× PCR buffer, 2.5 mM of each dNTP, and 10 pmol each of sense and antisense primers in a total volume of 50 µl. PCR conditions were as follows: 94°C for 30 s, 56°C for 30 s, and 72°C for 1 min (for 35 cycles). Moreover, the mouse housekeeping gene ribosomal protein large P0 (Rplp0), as a control, was amplified by RT-PCR with specific primers (sense primer, CGACCTGGAAGTCCAACTA; antisense primer, ATCTGCTGCATCTGCTTG) and Ex Taq polymerase (TaKaRa). PCR conditions for Rplp0 were as follows: 94°C for 30 s, 50°C for 30 s, and 72°C for 30 s. The PCR products were analyzed by electrophoresis on 2% agarose gels, stained with ethidium bromide, and visualized by ultraviolet irradiation.
Preparation of the protein and western blot analysis of hFGF2
Protein from the cells was isolated using the PRO-PREP protein extraction kit (iNtRON Biotechnology, Seoul, Korea) according to the manufacturer's instructions. The proteins from cell culture media were isolated by the trichloroacetic acid (TCA) method as follows: one volume of 100% (w/v) of TCA stock was added to four volumes of the cell culture medium, and incubated for 10 min at 4°C. Then the sample was centrifuged at 17,000 g for 5 min, and the supernatant was discarded. The pellet was washed five times with cold acetone and dried by placing the tube in a 95°C heat block for 10 min to evaporate acetone. For sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the pellet was dissolved in 1× sample buffer. For western blots, equivalent amounts of protein were separated on a 12% SDS-PAGE gel and were transferred to polyvinylidene fluoride membranes (Bio-Rad Co., Hercules, CA, USA). The membranes were blocked with 5% skimmed milk and 0.05% Tween 20 in Tris-buffered saline (TBS) overnight at 4°C and then blotted with a mouse monoclonal anti-bFGF antibody (1:1000 dilution; Sigma, F6162) or a mouse anti-β-actin antibody (1:2000 dilution; Santa Cruz Biotechnology, sc-47778) overnight at 4°C. After three washes with 0.05% Tween 20 in TBS, the membranes were incubated for 1 h with a goat anti-rabbit IgG-horseradish peroxidase conjugate (1:2000 dilution; Santa Cruz, sc-2005). The membranes were then washed three times with 0.05% Tween 20 in TBS. The protein bands were detected by incubation with enhanced-chemiluminescence agents (Amersham Co., Arlington Heights, IL, USA).
Preparation of donor cells
To prepare donor cells, we thawed gene-targeted cells and plated them in a culture dish containing the donor cell culture medium; we cultured the cells for 2−3 days. On the day of the experiment, donor cells were harvested using the TrypLE solution, washed three times, and treated with 3 mg/ml protease for 50 s at room temperature. Donor cells were then washed three times and resuspended in the donor cell preparation medium [TCM199-HEPES (Gibco) supplemented with 0.2 mM sodium pyruvate].
Preparation of recipient oocytes
The preparation of recipient oocytes was conducted according to previously reported methods (Kim et al., Reference Kim, Park, Park, Noh, Noh, Park, Lee, Jeong, Riu and Park2012). To prepare recipient oocytes, bovine ovaries were obtained from a slaughterhouse and transported to the laboratory within 2 to 4 h in PBS at 35°C. Cumulus–oocyte complexes (COCs) were retrieved from antral follicles between 2 and 6 mm in diameter using an 18-gauge needle attached to a 10-ml syringe. The medium used for COCs collection was HEPES-buffered Tyrode's medium. Groups of 10 COCs were cultured in 50-μl droplets of the TCM-199 maturation medium (Gibco) supplemented with 10% FBS (Gibco), 0.2 mM sodium pyruvate, 1 μg/ml follicle-stimulating hormone (Follitropin-V; Bioniche Animal Health; Belleville, ON, Canada), 1 μg/ml estradiol-17β, and 25 μg/ml gentamicin. The droplets were covered with mineral oil and incubated at 38.8°C in an incubator with 5% CO2, 5% O2, and 90% N2 for 18–20 h. For enucleation, a metaphase II plate and the first polar body were visualized under an inverted microscope (Olympus; Tokyo, Japan) equipped with the Oosight™ spindle-check system (Cri, Hopkinton, MA, USA); the removal involved the squeezing method.
SCNT
The SCNT was conducted according to previously reported methods (Kim et al., Reference Kim, Park, Park, Noh, Noh, Park, Lee, Jeong, Riu and Park2012). A single knock-in donor cell was placed in the perivitelline space of the enucleated oocyte in the nuclear transfer medium [TCM199–HEPES containing 0.6% fatty acid-free (FAF) bovine serum albumin (BSA) and 10 μg/ml phytohemagglutinin] through the opening made during enucleation. Oocyte-knock-in donor cell couplets were placed in the cell fusion medium (0.3 M mannitol, 0.5 mM HEPES, 0.05 mM CaCl2, and 0.1 mM MgSO4) and subjected to an electrical pulse of 1.3 kV/cm for 20 μs (delivered using the Electro Cell Fusion Generator, Model LF101; NEPA#GENE; Chiba, Japan). After fusion, the reconstructed knock-in embryos were maintained in TCM199–HEPES supplemented with 20% FBS for 1 h, activated in 10 μM calcium ionophore for 5 min, and then exposed to 2 mM 6-dimethylaminopurine for 3 h. After activation, the reconstructed knock-in embryos were cultured in the CR1aa medium supplemented with 0.3% FAF-BSA for 2 days. They were then co-cultured for 6 days with the cumulus cells in the CR1aa medium containing 10% FBS, 1 μM EGF, and 1 μM insulin-like growth factor (IGF) at 38.8°C in an atmosphere of 5% CO2, 5% O2, and 90% N2.
PCR analysis of the cloned embryos
For PCR, one cloned embryo in a PCR tube was incubated in 10 µl of digestion buffer [50 mM Tris–HCl (pH 8.5), 1 mM ethylenediaminetetraacetic acid, 0.005% SDS, 200 µg/ml proteinase K] for 2 h at 55°C; the tube was incubated for 10 min at 95°C. After digestion, 10 µl of extract was used directly for PCR after addition of 2% Tween 20 (final concentration) to the PCR mixture. The first PCR was conducted under the same conditions as the 3′-arm PCR during the colony screening. Nested PCR was conducted using 2 µl of PCR products from the first PCR as a template with sense (GCCTGCTTGCCGAATATCATGGTGGAAAAT), antisense (CATGAAAGTGAAAAGTGAAAGGGAAGTC) primers and Go Taq DNA polymerase (Promega) as follows: pre-denaturation at 94°C for 2 min, denaturation at 94°C for 30 s, annealing at 59°C for 30 s, and extension at 72°C for 1 min (38 cycles from denaturation to extension), and final extension at 72°C for 5 min. The PCR products were confirmed by electrophoresis on a 1.2% agarose gel.
Results
Construction of the knock-in vector to produce the human FGF2 protein in the bovine β-casein gene locus using the F2A self-processing peptide
The diagram of the knock-in vector for expression of hFGF2 in the bovine β-casein locus is shown in Fig. 1. The knock-in vector consisted of the 5′-homologous arm, F2A sequence, hFGF2 cDNA, BGH polyA signal, PGKneo, 3′-homologous arm, and diphtheria toxin A fragment (DT-A) gene. The Neo gene and DT-A gene that did not contain a polyA signal were used as a positive selection marker and negative selection marker, respectively. In this study, the knock-in vector was designed using a positive–negative selection marker and a poly A trap strategy. The F2A-fused human FGF2 cDNA was introduced into the exon 3 locus of the bovine β-casein gene. The hFGF2 mRNA could be expressed together with the β-casein signal sequence from exons 2 and 3 owing to their linkage through the F2A sequence. If the knock-in vector underwent homologous recombination in the bovine β-casein gene locus, then hFGF2 was expressed under the control of the bovine β-casein promoter and all gene-regulatory sequences including an enhancer. The deduced nucleotide and amino acid sequence of the β-casein signal sequence/F2A/hFGF2 fusion gene is shown in Fig. 2. We analyzed the cleavage site in the β-casein signal sequence and in the β-casein signal sequence/F2A/hFGF2 fusion protein by using the SignalP 4.1 server (http://www.cbs.dtu.dk/services/SignalP/). This fusion protein is probably cleaved between positions 15 (alanine) and 16 (arginine) of the β-casein signal sequence (Fig. 2). The F2A self-cleavage site in the F2A sequence is located between positions 50 (proline) and 51 (glycine; Fig. 2). The F2A peptide is believed to disrupt translation and to impair normal peptide bond formation between the proline (position 50) and glycine (position 51) of F2A, without affecting translation of the hFGF2 protein (Fig. 2 and Fig. 3). According to this model, hFGF2 mRNA is probably translated with the bovine β-casein sequence and F2A peptide. After that, the human FGF2 protein can be cleaved to remove the bovine β-casein sequence and F2A peptide from the fusion protein (Fig. 3).

Figure 2 Structure of the hFGF2 gene fused with the bovine β-casein signal sequence through the F2A sequence. Deduced nucleotide and amino acid sequences of the fusion gene. Nucleotide residues are numbered on the right; amino acid residues are numbered on the left. Nucleotide 1 is base A of the initiator ATG codon in the bovine β-casein gene. Bold nucleotides indicate a signal sequence from exons 2 and 3 of the bovine β-casein gene. Underlining indicates the F2A sequence. Bold letters show the human FGF2 amino acid sequence. The open arrow indicates the cleavage site for the signal sequence in β-casein. The black arrow indicates a self-cleavage site in the F2A peptide during translation.

Figure 3 The diagram of transcription, translation, and cleavage of the human FGF2 protein. The hFGF2 gene in the knock-in vector was fused with F2A in exon 3 of the β-casein gene; the β-casein signal sequence (which leads the β-casein protein to the endoplasmic reticulum via the secretory pathway) was located in exon 2 of the β-casein gene. The F2A peptide is believed to disrupt translation and to impair normal peptide bond formation between the proline and glycine residues of F2A, without affecting translation of the hFGF2 protein. If the knock-in vector undergoes homologous recombination, then the altered β-casein gene produces hFGF2 mRNA using gene-regulatory DNA sequences including enhancers of the β-casein gene. The fusion proteins are translated by a ribosome and are translocated to the endoplasmic reticulum owing to the β-casein signal peptide. During the protein synthesis, the human FGF2 protein is released form the fusion protein and is translocated to the endoplasmic reticulum. The human FGF2 proteins may be secreted according to the endoplasmic reticulum and Golgi pathway.
Expression and secretion of hFGF2 by the knock-in vector in the mouse mammary gland HC11 cells
The knock-in vector contained the 5′-arm, which had a minimum promoter including a TATA box, and human F2A-fused hFGF2 cDNA was inserted into exon 3 of the β-casein gene. Therefore, we analyzed the expression of hFGF2 using RT-PCR and western blotting in the HC11 mouse mammary epithelial cells transfected with the knock-in vector to confirm expression of human FGF2 in this knock-in vector.
Using RT-PCR, the mRNA of hFGF2 fused to β-casein exon 2 and F2A was detected in the HC11 cells transfected with the knock-in vector (using the sense primer located in β-casein exon 2 and the antisense primer located in hFGF2; Fig. 4 A). Moreover, the hFGF2 protein was expressed in the HC11 cells transfected with the knock-in vector, and more hFGF2 protein was secreted into the culture medium than in control HC11 cells (Fig. 4 B).

Figure 4 Expression and secretion of human FGF2 protein into the cell culture medium. We used HC11 cells transfected with the knock-in vector. (A) mRNA expression of the bovine β-casein gene that was fused to the human FGF2 gene was detected by RT-PCR using specific primers in the HC11 cells transfected with the knock-in vector. (B) Western blot analysis of human FGF2 protein in the cell culture medium and in cells. ‘C’ means HC11 cells serving as a negative control; ‘KI’ means HC11 cells transfected with the knock-in vector.
These results indicate that hFGF2 cDNA as a transgene was expressed by the β-casein gene-regulatory sequences in the knock-in vector, and the hFGF2 protein was secreted into the cell culture medium after the β-casein signal sequence and F2A peptide were chopped off.
Evaluation of CRISPR/Cas9 in bovine MAC-T cells
To develop knock-in fibroblasts in this study, we used the CRISPR/Cas9 system. The sgRNA target sequence (CTGGAAGAACTCAATGTACCTGG) was located in the β-casein exon 2 (Fig. 5 A). To test whether the CRISPR/Cas9 system could be used for targeted disruption of an endogenous gene in bovine cells, we analyzed genomic DNA isolated from MAC-T cells transfected with sgRNA and the Cas9 expression vector using T7 endonuclease I (T7E1), an endonuclease that specifically cleaves heteroduplexes formed by hybridization of wild-type and mutant DNA sequences. We found that mutations were induced only when the cells were co-transfected with both sgRNA and the Cas9 expression vector (Fig. 5 C). Next, we are analyzed the CRISPR/Cas9 system using the target or non-target RFP–GFP reporter system in the MAC-T cells to test whether the sgRNA and Cas9 can cleave the target sequence incorporated between the RFP and GFP sequences. In this reporter, the GFP sequence is fused to the RFP sequence out-of-frame. GFP is expressed only when the target sequence is cleaved by a site-specific nuclease, which causes small, frame-shifting insertions or deletions around the target sequence by means of error-prone nonhomologous end-joining repair of double-stranded breaks. We found that GFP expression was only detected in the RFP-expressing cells when the cells were cotransfected with target RFP–GFP reporter, sgRNA and the Cas9 expression vector (Fig. 5 B, D).

Figure 5 The target site of sgRNA and validation of the sgRNA/Cas9 cleavage. (A) The target site of sgRNA in the bovine β-casein gene exon 3. (B) A schematic overview of the cell base assay using an RFP–GFP reporter gene. (C) The T7 endonuclease I assay in the MAC-T cells transfected with sgRNA/Cas9. (D) RFP and GFP expression of Mac-T cells transfected with reporter gene and sgRNA and Cas9 plasmid. The top panel shows only RFP expression by transfection of non-target reporter, sgRNA and Cas9 expression vector. The middle panel shows only RFP expression by transfection of target reporter and Cas9 expression vector. The lower panel shows RFP–GFP expression by transfection of target reporter, sgRNA and Cas9 expression vector. M, size markers; NC, negative control; sgRNA/Cas9, Mac-T cell transfected with the sgRNA/Cas9 expression vector.
Efficiency of the knock-in in bovine fibroblasts by means of the CRISPR/Cas9 system
Results from the knock-in experiment with CRISPR/Cas9-assisted homologous recombination of the knock-in vector are presented in Table 1. In total, 209 and 62 Neo-resistant colonies in male cells, transfected with or without the CRISPR/Cas9 system and knock-in vector, respectively, were analyzed by PCR. In total, 20 colonies underwent homologous recombination in the fibroblasts co-transfected with the CRISPR/Cas9 system and the knock-in vector. Furthermore, only two colonies were confirmed as knock-in colonies in the fibroblasts transfected with the knock-in vector alone. In female cells, 104 of 130 colonies co-transfected with the CRISPR/Cas9 system and knock-in vector were validated as test positive by PCR. In addition, after transfection of the knock-in vector alone, one colony was identified as test positive. Homologous recombination occurred more frequently in the fibroblasts (32.2 and 80%) cotransfected with the CRISPR/Cas9 system and knock-in vector compared to the knock-in vector alone (0.9 and 1.55%, respectively). Figure 6 A presents the PCR results for a representative group of G418-resistant colonies. Only the expected 2.5-kb PCR products were obtained when we tested the knock-in colonies using the Neo 3–4 and FGF KI Sc AS3 primer located outside the 3′-recombination region (top panel; Fig. 6 A). In addition, 6.7-kb PCR products were successfully amplified using the pβC5′-Sc S3 primer located outside the 5′-recombination region and the Neo 5–2 primer (middle panel; Fig. 6 A). Therefore, targeted and non-targeted alleles were detected in the knock-in colonies using the βE2 5′-sense 1 primer located in exon 2 and the FGF KI Sc AS3 primer located outside the 3′-recombination region. All knock-in fibroblasts were identified as biallelic knock-in fibroblasts; this result suggested that homologous recombination occurred only in one allele of the β-casein according to PCR (lower panel; Fig. 6 A). Therefore, to further confirm precise targeting at the bovine β-casein gene locus, we carried out Southern blot analysis using bovine β-casein exon 7 probe in the knock-in colonies. The targeted and wild-type allele was detected with 8.3 kb and 6.4 kb band, respectively. Southern blot analysis using neo probe revealed that four of the 13 colonies had random integration of knock-in vector in these colonies (Fig. 6 B). This finding indicates that the use of the CRISPR/Cas9 system in the knock-in technology leads to an increased incidence of homologous recombination in fibroblasts.
Table 1 Frequencies of knock-in in the β-casein gene locus in bovine fibroblasts


Figure 6 Analysis of knock-in fibroblasts and blastocysts by PCR. (A) PCR analysis of the knock-in fibroblasts. The primer set presented in the targeted locus map. The top panel shows the 3′-arm PCR using neo 3–4 and FGF KI Sc AS3 primers located outside the 3′-recombination region. The middle panel shows the 5′-arm PCR using the pβC5′-Sc S3 primer located outside the 5′-recombination region and the Neo 5–2 primer. The lower panel shows targeted (5.6 kb) and non-targeted (3.5 kb) alleles using βE2 5′-S1 primer located in exon 2 and the FGF KI Sc AS3 primer located outside the 3′-recombination region. (B) Southern blot analysis of knock-in colonies with exon 7 probe (upper panel) located outside the 3′-recombination region and neo probe (lower panel) located targeting vector as inner probes. The 8.3 kb and 6.3 kb band in the upper panel indicated targeted and wild-type alleles, respectively. The 8.3 kb band in the lower panel indicated targeted alleles. (C) PCR analysis of knock-in blastocysts. BL indicates control blastocysts and KI-BL indicates knock-in blastocysts produced via SCNT from knock-in fibroblasts. PC, positive control; NC, negative control; M, size markers; Number, colony or blastocyst sample number. (D) Cloned day-2 (1 and 2) and day-8 (3 and 4) embryos produced from normal ear cells (1 and 3) and knock-in FGF cells (2 and 4). Bar, ×200 magnification.
Generation and development of knock-in cloned embryos in vitro
The results of in vitro development of cloned embryos produced from two donor nuclei (normal ear cell and knock-in hFGF2 cell) are shown in Table 2 and Fig. 6 C. On day 2 after activation, the cleavage rates of embryos reconstructed from the fused oocytes were 81.7% (196/240, Fig. 6 D-1) and 80.3% (183/228, Fig. 6 D-2) for the ear cell group and knock-in hFGF2 cell group, respectively, and the results were not significantly different. However, on day 8 after activation, the blastocyst development rates were significantly lower in the knock-in cell group (25.7%, 47/183, Fig. 6 D-3) than in the control ear cell group (34.7%, 68/196, Fig. 6 D-4; P < 0.05).
Table 2 The in vitro development of cloned embryos from normal and gene-targeted fibroblasts (n = 6)

*Means with different superscripts in the same column are statistically significantly different ( a,bP < 0.05).
To confirm the knock-in in the blastocysts, we conducted direct PCR analysis using knock-in and control blastocysts. PCR products were successfully amplified only in the knock-in blastocysts and not in control blastocysts (Fig. 6 C). Thus, these data suggest that we produced knock-in blastocysts (by means of gene targeting) that could be used to develop transgenic cattle that express the hFGF2 gene under the β-casein gene-regulatory sequences.
Discussion
In this study, knock-in fibroblasts and blastocysts, which may be used to produce a mammary-gland bioreactor expressing the hFGF2 gene, were generated by gene targeting of the β-casein gene exon 3 locus with insertion of F2A-fused hFGF2 cDNA. In these knock-in fibroblasts, hFGF2 is expressed owing to endogenous gene-regulatory DNA sequences including all enhancers of the bovine β-casein gene.
In the past several years, there has been significant interest in the development of transgenic animals to produce recombinant proteins for medicinal purposes. However, it has been very difficult to increase the production level in transgenic animals. In the past, many transgenic animals have been produced by microinjection, virus vector-mediated gene transfer, sperm-mediated gene transfer, or nuclear transfer methods (Houdebine, Reference Houdebine2000, Reference Houdebine2009; McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000). Microinjection is a method involving injection of a foreign gene into the pronucleus of a fertilized egg using a micromanipulator for expression of a foreign gene; this is the most widely used method (Clark, Reference Clark1998; Houdebine, Reference Houdebine2000, Reference Houdebine2009; Robl et al., Reference Robl, Wang, Kasinathan and Kuroiwa2006). The production efficiency of transgenic animals based on microinjection is only 2–3%, and this method is difficult to apply to domestic animals because the genes are integrated at random sites throughout the host animal genome and usually have low levels of expression ( Clark, Reference Clark1998; Houdebine, Reference Houdebine2000; Robl et al., Reference Robl, Wang, Kasinathan and Kuroiwa2006; Houdebine, Reference Houdebine2009). In addition, virus vector-mediated gene transfer and sperm-mediated gene transfer result in random integration, similar to microinjection. Thus, it is not possible to use this method to insert a foreign gene into a specific target site or to eliminate expression of a specific endogenous gene by homologous recombination.
Generation of transgenic domestic animals via nuclear transfer using transfected cells provides a number of advantages over the classic DNA microinjection technique. One major advantage of the former is that integration and expression of the transgene can be evaluated in the transfected cells before they are used as a nuclear donor (McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000). Furthermore, transgenesis by nuclear transfer offers a strategy for either random or targeted genetic modification of domestic animals. Gene-targeted sheep created by nuclear transfer were generated using ovine fetal fibroblasts where researchers inserted the α1-antitrypsin gene into the α1(I) procollagen locus (McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000). Using this method, these authors showed that transgenic sheep could be hormonally induced to lactate and achieve high expression (650 μg/ml) of α1-antitrypsin in their milk. These results indicate that gene targeting is a powerful method for production of animal bioreactors. Nevertheless, the authors used the β-lactoglobulin promoter for expression of the α1-antitrypsin gene in the α1(I) procollagen locus of sheep. Although the α1-antitrypsin gene was driven by the β-lactoglobulin promoter, the expression was not induced using gene-regulatory DNA sequences because of the use of the transgenic promoter (McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000). Recent work has demonstrated the importance of distal enhancers for transcriptional gene regulation and tissue-specific gene expression (Heintzman et al., Reference Heintzman, Hon, Hawkins, Kheradpour, Stark, Harp, Ye, Lee, Stuart, Ching, Ching, Antosiewicz-Bourget, Liu, Zhang, Green, Lobanenkov, Stewart, Thomson, Crawford, Kellis and Ren2009; Visel et al., Reference Visel, Blow, Li, Zhang, Akiyama, Holt, Plajzer-Frick, Shoukry, Wright, Chen, Afzal, Ren, Rubin and Pennacchio2009).
In this study, we inserted hFGF2 into exon 3 of the β-casein gene locus linked to the F2A sequence in the knock-in vector in order to express hFGF2 using gene-regulatory DNA sequences including all enhancers of the bovine β-casein gene. If this knock-in vector undergoes homologous recombination in the cell, then the human FGF2 gene can probably be expressed as well as the endogenous bovine β-casein under the influence of the gene-regulatory DNA sequences including all enhancers of the bovine β-casein gene. In addition, the human FGF2 gene is fused with the F2A sequence into exon 3 of the β-casein gene locus; the signal sequence for secretion of β-casein is known to be present in exon 2 of the β-casein gene. The F2A peptide is cleaved off by the self-cleaving system during protein synthesis (Szymczak & Vignali, Reference Szymczak and Vignali2005). In the present study, by means of RT-PCR and western blot analysis, we detected mRNA and protein expression of hFGF2 in HC11 mouse mammary epithelial cells, which randomly integrated the knock-in vector into the genome. In addition, the human FGF2 protein was secreted into the cell culture medium more abundantly compared with control cells. Also, human FGF2 protein was detected in the control cells. It is thought that monoclonal anti-human FGF2 antibody used in this study may cross-react with mouse FGF2 in the cells. Because the promoter in the 5′-arm of the knock-in vector used in this study may contain basic transcriptional components, the human FGF2 gene was driven by a minimal promoter including the TATA box motif. These results show that hFGF2 proteins in knock-in cells could be cleaved to remove the β-casein signal sequence and F2A peptide and to promote secretion of the hFGF2 protein through the endoplasmic reticulum and Golgi pathway.
In this study, we created knock-in somatic cells, which may be used to produce human FGF2 in the mammary gland, by gene targeting of the β-casein gene locus using the CRISPR/Cas9 system-mediated homologous recombination of the knock-in vector. Previously, transgenic sheep were created by gene targeting (nuclear transfer) to produce a recombinant protein in the goat mammary glands (McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000). In that work, the researchers used a positive selection marker (Neo gene) to select the knock-in somatic cells. Using this approach, they obtained very high targeting efficiency (66%). However, the targeting frequency is generally known to be very low in somatic cells when conventional methods are used (Denning & Priddle, Reference Denning and Priddle2003; Laible & Alonso-González, Reference Laible and Alonso-González2009).
Recently, new technologies such as the ZFN, TALEN, and CRISPR/Cas9 systems were used to create gene knockout animals. In the CRISPR/Cas9 system, high DNA cleavage activity was reported when two components of sgRNA/Cas9 were introduced into the cultured cells, mice, rats, or domestic animals (Porteus & Carroll, Reference Porteus and Carroll2005; Geurts et al., Reference Geurts, Cost, Freyvert, Zeitler, Miller, Choi, Jenkins, Wood, Cui, Meng, Vincent, Lam, Michalkiewicz, Schilling, Foeckler, Kalloway, Weiler, Ménoret, Anegon, Davis, Zhang, Rebar, Gregory, Urnov, Jacob and Buelow2009; Cui et al., Reference Cui, Ji, Fisher, Wu, Briner and Weinstein2011; Wei et al., Reference Wei, Liu, Yu, Zhang, Gao and Jiao2013; Sung et al., Reference Sung, Jin, Kim and Lee2014). These results suggest that it is possible to achieve CRISPR/Cas9-mediated homologous recombination for a knock-in of an exogenous transgene into a specific gene locus to ensure stable expression of the transgene. In the present study, we show that the CRISPR/Cas9 system can induce homologous recombination with very high efficiency (75.4–80%) in bovine primary fibroblasts. This homologous recombination efficiency is much higher than that reported previously (McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000; Denning & Priddle, Reference Denning and Priddle2003; Laible & Alonso-González, Reference Laible and Alonso-González2009). Efficient genetic modification of the fibroblasts for SCNT is very important to produce transgenic domestic animals (via knock-in) serving as an animal bioreactor because embryonic stem cells in domestic animals have not been established. Knock-in using CRISPR/Cas9-mediated homologous recombination of donor DNA has not been reported in bovine fibroblasts. Our results indicate that a knock-in using CRISPR/Cas9-mediated homologous recombination of a knock-in vector (donor DNA) in bovine fibroblasts is a very powerful technique for production of knock-in somatic cells for SCNT.
In this study, we created knock-in blastocysts by SCNT using knock-in fibroblasts and we detected the knock-in vector in the knock-in blastocysts by PCR. These results indicate that knock-in blastocysts were successfully produced using knock-in fibroblasts, even though the development rate of blastocysts was lower compared with control fibroblasts. It is known that production of transgenic fibroblasts requires long-term in vitro culture for gene transfer and selection of transgenic fibroblasts with antibiotics (McCreath et al., Reference McCreath, Howcroft, Campbell, Colman, Schnieke and Kind2000; Song et al., Reference Song, He, Hua, Lan, Liu, Cheng, Zhang, Li, He, Liu and Zhang2011; Cheng et al., Reference Cheng, Yu, Liu, Deng, Ma and Wang2013; Bressan et al., Reference Bressan, Miranda, Bajgelman, Perecin, Mesquita, Fantinato-Neto, Merighe, Strauss and Meirelles2013). Development of blastocysts using SCNT with long-term-cultured transgenic fibroblasts is generally less productive compared to nontransgenic fibroblasts in cattle (Song et al., Reference Song, He, Hua, Lan, Liu, Cheng, Zhang, Li, He, Liu and Zhang2011). However, cloned cattle were produced from fibroblasts after long-term culture, and the gene-targeted cattle were developed by means of SCNT using targeted fibroblasts after long-term culture (Kubota et al., Reference Kubota, Yamakuchi, Todoroki, Mizoshita, Tabara, Barber and Yang2000; Kuroiwa et al., Reference Kuroiwa, Kasinathan, Matsushita, Sathiyaselan, Sullivan, Kakitani, Tomizuka, Ishida and Robl2004). These reports indicate that the knock-in fibroblasts created in our study could be used for production of knock-in cattle.
In the present study, we generated knock-in fibroblasts by CRISPR/Cas9-mediated homologous recombination of a knock-in vector, which contained 5′- and 3′-arms with a homologous sequence and human FGF2 as a transgene, in bovine fibroblasts. We also confirmed that the human FGF2 gene was expressed in mouse mammary gland HC11 cells and that FGF2 protein was secreted into the cell culture medium. However, we created the knock-in blastocysts by SCNT using knock-in somatic cells. In conclusion, we show for the first time that the CRISPR/Cas9-mediated homologous recombination can be efficiently accomplished in bovine fibroblasts. The resulting knock-in cells and blastocysts could be used for the production of transgenic cattle that express hFGF2 within the bovine β-casein gene locus.
Acknowledgements
This project was supported by a grant from the Next-Generation BioGreen 21 Program (No. PJ011176 and PJ009075), Rural Development Administration, Republic of Korea.
We gratefully acknowledge Feng-Qi Zhao (Department of Animal Science, College of Agriculture and Life Sciences, University of Vermont, Burlington, USA) for the generous gift of MAC-T cells for the cell culture study.