


Introduction
The scale insects (Hemiptera: Coccoidea) are a major group of pests on crops and ornamental plants worldwide. They include more than 7700 described species in 1050 genera (Ben-Dov et al., Reference Ben-Dov, Miller and Gibson2006) belonging to 31 extant and 14 fossil families (Kondo et al., Reference Kondo, Gullan and Williams2008). Nearly all scale insects can be identified only morphologically to a species level by examining characters present in adult females. However, even when adult females are examined, it is sometimes difficult to distinguish between very similar species and determining if slight differences represent inter- or intra-specific variation. Studies of sequence variation in several gene regions have been carried out in an attempt to resolve this problem. Initial efforts examined three nuclear genes: the D2 region of 28S rDNA, elongation factor 1α (Downie & Gullan, Reference Downie and Gullan2004; Morse & Normark, Reference Morse and Normark2006) and ITS (Beuning et al., Reference Beuning, Murphy, Wu, Batchelor and Morris1999; Park et al., Reference Park, Leem, Hahn, Suh, Hong and Oh2010b). ITS sequences were able to discriminate closely related species, but the high incidence of indels made sequence alignment difficult except for closely allied taxa. The 28S sequences were easier to align but lacked enough variation to resolve some species. Another investigation examined sequence variation in the 16S–23S rDNA of the vertically transmitted endosymbiotic microbe, Tremblaya princeps (Thao et al., Reference Thao, Gullan and Baumann2002; Baumann & Baumann, Reference Baumann and Baumann2005), as a tool for scale insect identification, but it too lacked enough variation. A more recent study tested the possibility of identifying mealybug species through the analysis of four markers: two nuclear genes, one mitochondrial gene and one gene from the endosymbiont (Malausa et al., Reference Malausa, Fenis, Warot, Germain, Ris, Prado, Botton, Vanlerberghe-Masutti, Sforza, Cruaud, Couloux and Kreiter2010). The standard barcode region of cytochrome c oxidase subunit I (COI) gene was targeted for analysis in this study, but the standard primer set (Folmer et al., Reference Folmer, Black, Hoeh, Lutz and Vrijenhoek1994) only generated an amplicon in a few species. It proved possible to recover more species with a new primer set that amplified a 491 bp segment of the barcode region, but these records are too short to qualify for recognition as formal barcodes. Nonetheless, the results were promising enough to provoke further investigation.
DNA barcoding aims to identify species through the analysis of sequence variation in a standardized DNA sequences. A 658 bp segment near the 5′ end of the mitochondrial COI has been accepted as the core barcode region for the animal kingdom (Hebert & Gregory, Reference Hebert and Gregory2005; Hebert et al., Reference Hebert, Ratnasingham and deWaard2003b). Many studies have now established that this region possesses enough sequence variation to discriminate most animal species (Hajibabaei et al., Reference Hajibabaei, Janzen, Burns, Hallwachs and Hebert2006; Hebert et al., Reference Hebert, Cywinska, Ball and deWaard2003a). However, in certain groups, such as scale insects, it has proven difficult to amplify the barcode region, suggesting mismatches between the primers and the target DNA. A recent study confirmed this fact for certain scale insects, revealing two substitutions at critical positions near the 3′ binding site for the forward primer. This observation allowed the design of a new forward primer, which showed high success when tested on a few species of scale insects (Park et al., Reference Park, Suh, Oh and Hebert2010a). The present study examines the utility of this primer set in a more comprehensive way, by testing its performance on 88 species from the two most diverse families of scale insects, the Pseudococcidae (mealybugs) and the Diapsidae (armored scales). Aside from testing amplification success, this investigation considers both the utility of the resultant sequences in scale insect identification and certain unusual properties of these sequences.
Materials and methods
Specimens
Most of the specimens examined in this study were obtained from collections made by the National Plant Quarantine Service in South Korea. However, specimens of Planococcus ficus were provided by the Plant Protection Research Institute of South Africa, and some Planococcus kraunhiae and Crisicoccus matsumotoi were contributed by the Shimane Agricultural Technology Center, Japan. In total, 524 specimens were analyzed, representing 88 species from two families of scale insects. Whenever possible, at least five specimens were analyzed of each species from each of the localities where it was collected.
DNA extraction, PCR and sequencing
DNA was extracted from most individuals using a standard glass fiber extraction protocol (Ivanova et al., Reference Ivanova, deWaard and Hebert2006); the remainder (9.4%) were extracted using a DNeasy kit (Qiagen, Hilden, Germany) following the manufacturer's protocol, except that the final elution step was performed with 60 μl of distilled water instead of 200 μl buffer. Specimen cuticles were recovered after DNA extraction and then stored in ethanol until slide mounted. All of the vouchers from this study are deposited in the scale insect collection of National Plant Quarantine Service (NPQS), Busan, Korea. A 649-bp segment of the barcode region was amplified from most specimens using the primer PcoF1-5′CCTTCAACTAATCATAAAAATATYAG3′ (Park et al., Reference Park, Suh, Oh and Hebert2010a) and LepR1-5′TAAACTTCTGGATGTCCAAAAAATCA3′. A few specimens were amplified using two alternate primer sets (LepF1/LepR or C_tRWF/LepR1) before the PcoF1 primer was developed. PCRs were performed using a Maxime® PCR PreMix (iNtRON Biotechnology, Seongnam, Korea) with 2.0 ρmol of each primer and 2–50 ng of template DNA in a 20 μl reaction, or a standard 12.5 μl reaction developed by Ivanova et al. (Reference Ivanova, deWaard and Hebert2006). PCR thermocycling was done under the following conditions: 2 min at 95°C; five cycles of 40 s at 94°C; 40 s at 45°C, 60 s at 72°C; 40 cycles of 40 s at 94°C; 40 s at 51°C; 60 s at 72°C; 5 min at 72°C; held at 4°C. PCR products were visualized in a 2% agarose gel stained with ethidium bromide and bidirectionally sequenced using a BigDye Terminator ver. 3.1 Cycle Sequencing Kit (Applied Biosystems Inc., Foster, CA) on an ABI 3730XL capillary sequencer. Contigs were assembled using CodonCode Aligner ver 3.5.6 (CodonCode Co., Dedham, MA) and were subsequently aligned using the same software or MEGA (Tamura et al., Reference Tamura, Dudley, Nei and Kumar2007). All sequences were deposited in GenBank (Accession numbers HM474068–HM474409), while the collection data are available in the DBSI project file in BOLD (http://www.barcodinglife.org). Sequence divergences were calculated using the Kimura 2-parameter (K2P) model (Kimura, Reference Kimura1980), and a neighbour-joining (NJ) tree was generated by MEGA or by the Taxon ID tree function in BOLD (Ratnasingham & Hebert, Reference Ratnasingham and Hebert2007).
Results
A DNA barcode record, almost 649 bp in length, was recovered from 343 of 524 specimens, a 65% success rate. These sequences were derived from 75 of the 88 species and included members of 32 genera. So, the success in barcode recovery per species level exceeded 90% for mealybug and 80% for armored scales. We recovered 30 additional sequences from seven species of mealybugs that possessed a 1 or 2 bp indel, suggestive of pseudogenes. These cases, which involved the deletion of one or two thymine residue deletions, were detected in every individual of seven mealybug species (Dysmicoccus wistariae, Ferrisia malvastra, Ferrisia virgata, Palmicultor lumpurensis, Pseudococcus calceolariae, Pseudococcus jackbeardsleyi, Pseudococcus viburni and an unidentified Pseudococcus sp.). These deletions all occurred at the same position (149–150) in the standard COI barcode region although an additional deletion was also present at position 476 in all seven individuals of P. jackbeardsleyi (S2). These sequences are included in the compressed phylogenetic tree (fig. 1) and were submitted to GenBank with a pseudogene notation.

Fig. 1. Distribution of intra-specific genetic distances (K2P) at COI 5′ region for 343 individuals of 68 species of scale insects. Mean K2P distance of a species was calculated using the sequence analysis in BOLD and percentage of species within certain intra-specific value was represented.
The G·C content of the COI gene regions from the scale insects examined in this study are very low, averaging just 16.4%. Species of mealybugs have a lower G·C content (14.9%) than those of armored scales (18.2%), but there was significant variation in G·C content among the members of each family (table 2). The mealybugs Atrococcus paludinus and Paracoccus marginatus have the lowest values (12.6% and 12.9%, respectively), while the armored scales (Odonaspis secrata and Pseudaonidia duplex) have relatively high G·C content (30.2% and 33.5%, respectively). Mean G·C content was 18.2% and 28.4% in armored scales and 15.9% and 26.1% in mealybugs at codon positions 1 and 2, respectively, but just 8.2% in armored scales and 4.2% in mealybugs at position 3.
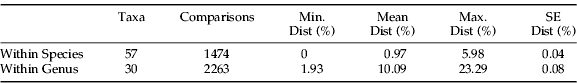
Figure 1 provides a compressed K2P/NJ tree of the patterns of sequence divergence among the 373 specimens with a barcode record while the entire tree is available in S3. Sequence divergences (K2P distance) between congeneric species averaged 10.09%, a value which was tenfold higher than the mean intra-specific variation (table 1). Some overlap was observed in the distribution of K2P distances among conspecific individuals (range 0–5.98%) and among congeneric species (range 1.93–23.29%). Although most species showed less than 1.0% divergence, 25% of them showed over 2.0% divergence (fig. 2), cases that suggest several overlooked species. Particularly deep divergences (2.5–4.6%) were found within three armored scale species (Lopholeucaspis japonica, Aspidiotus excises and Pseudaulacaspis pentagona). Six species of mealybug (Crisicoccus matsumotoi, Maconellicoccus hirsutus, Phenacoccus aceris, Phenacoccus solani, Planococcus ficus and Pseodococcus longispinus) also showed deep divergences (2.5–5.8%) (fig. 3).

Fig. 2. A compressed NJ tree of COI sequence divergences (K2P) in 75 mealybug and armored scale species. The number of specimens is indicated following each species name. ♦, sequences which have internal thymine residue deletion.

Fig. 3. NJ analysis (K2P) showing intra-specific variation in the barcode region for seven species of scale insects.
Table 1. Genetic divergences (K2P) among members of a species and between congeneric species of scale insects.
Discussion
DNA barcoding has gained increasing usage for the discrimination of animal species, but its application has proven difficult for certain groups, such as scale insects, because of inconsistent PCR amplification of the target COI 5′ region. Park et al. (Reference Park, Suh, Oh and Hebert2010a) recently developed a new forward primer (PcoF1), which, when combined with a standard reverse primer (LepR1), successfully amplified several scale insect species. This study involved a larger-scale test examining barcode recovery in 88 species of mealybugs and armored scales. Barcodes were recovered from 343 of 524 specimens (71%), a lower success rate than in the earlier study (92%) (Park et al., Reference Park, Suh, Oh and Hebert2010a). We suspect that this difference reflects the use of alternate DNA extraction methods because we had 58% success for 190 armored scale specimens and 76% success for 285 mealybug specimens using a Silica-based extraction method (Ivanova et al., Reference Ivanova, deWaard and Hebert2006) but 95% success when DNA was extracted using the DNeasy kit (ten of ten armored scales and 37 of 39 mealybugs), a success rate similar to that reported by Park et al. (Reference Park, Suh, Oh and Hebert2010a).
Park et al. (Reference Park, Suh, Oh and Hebert2010a) reported that scale insects have a characteristic deletion of three amino acids in the barcode region. The same deletion was detected in all 75 species examined in this study, suggesting that this deletion is shared by many, if not all, species in at least these two families of scale insects. The present study revealed one other anomaly; seven of the 40 mealybug species belonging to three different genera had a deletion of 1–2 thymine residues at position 149–150 in the COI 5′ barcode region (fig. 2). Because these deletions induced a frame shift mutation which disrupts translation of the COI protein, we expect that these sequences reflect the recovery of a NUMT. If so, the conserved location of the deletion suggests that all cases reflect an ancient transposition event from the mitochondrion to the nucleus that occurred before diversification of the three genera that now carry it. Given this shared ancestry of lineages carrying the NUMTs, one would expect them to form a cohesive cluster in the NJ tree, but they do not (fig. 2). Alternatively, deletions at this position may have evolved recurrently after nuclear transposition, reflecting an inherent tendency toward the loss of a thymine residue in a poly-T region of this gene in scale insects. Interestingly, a thymine insertion was detected in a species of Coelostomidiidae, another family of scale insects, at the same position (Kaur, M., personal communication). However, it remains important to rule out the possibility that these sequences represent the authentic mitochondrial sequence with the translation mechanism adjusting the reading frame. The recovery of a full mitochondrial genome sequence from these species is required to rule out the possibility that these deletions occur in the mitochondrial genome itself.
The G·C content of the barcode region of scale insects is very low, averaging just 16.4% versus typical values of 33–53% in other animal lineages (Min & Hickey, Reference Min and Hickey2007). As expected, the low G·C frequency is more pronounced at the third codon position (8.2% for armored scales, 4.2% for mealybugs), than at the first and second positions (18.2% and 28.4% in armored scales versus 15.9% and 26.1% in mealybugs) (table 2). The marsh mealybug, A. paludinus, has no G·C at the third position in the standard barcode region, while the papaya mealybug, P. marginatus, has only a single G·C base. The average G·C content (14.9%) for the barcode region in mealybugs (Pseudococcidae) is the lowest value for any insect family, while the 12.6% G·C content for A. paludinus is the lowest value known for any animal species. These values are extreme even when viewed from the context of bacteria genomes whose G·C contents range from 17–75%. The lowest bacterial values occur in species with small genome sizes (Andersson & Kurland, Reference Andersson and Kurland1998), especially the endosymbionts of insects, such as aphids and scale insects, which possess G·C contents of 17–33% (Moran & Wernegreen, Reference Moran and Wernegreen2000; Perez-Brocal et al., Reference Perez-Brocal, Gil, Ramos, Lamelas, Postigo, Michelena, Silva, Moya and Latorre2006; Moran et al., Reference Moran, McCutcheon and Nakabachi2008). Interestingly, a low G·C content (23–27%) also exists in the barcode region of aphids, animals that feed, like scale insects, on plant sap, a diet which is very deficient in organic nitrogen. Might the low G·C content in aphids, scale insects and their endosymbiotic bacteria represent an evolutionary adaptation to nitrogen scarcity because less nitrogen is required for an A·T than G·C pair?
Table 2. Variation in G·C content for the barcode region of COI in two families of scale insects.
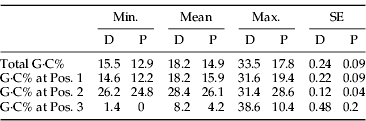
D, diaspididae; P, pseudococcidae.
We detected deep sequence divergence (>2.0%) in nine of the 75 species that we examined (fig. 2). Our analyses revealed that specimens of a single species from varied geographic regions often showed substantial genetic divergence, possibly reflecting cryptic species overlooked by current taxonomic treatments (fig. 3). However, we also detected deep intra-specific divergences in some cases where specimens were collected in close proximity. For example, P. aceris included two distinctive clusters with 5.6% divergence, and these specimens were all collected on south-western islands in the Korean Peninsula (fig. 3b). Interestingly, the specimens in one cluster were collected on Eriobotra trees on the island of Bogil-do, while specimens in the second cluster were collected from three other tree genera (Liquidamber, Zelkova and Sorbus) on another island, Wan-do. A second case of intra-specific variation involved C. matsumotoi, an important pest of pears in South Korea. Specimens of this species were obtained from three countries (South Korea, Japan and Canada). The sole Canadian specimen was closely similar to some Korean specimens, while the three Japanese specimens had slightly divergent sequences (fig. 3a). However, this species included two clusters with an intra-specific K2P distance of 2.5–4.0%. We also found two Pseudococcus species (comstocki and logispinus) that possessed two very genetically divergent lineages that could not be separated morphologically (fig. 2). To clarify these potential cases of cryptic species, more extensive sampling followed by detailed phenotypic and molecular studies will be required.
This study has established the feasibility of creating a comprehensive barcode library for scale insects. It has shown that this effort will reveal taxonomic situations worthy of deeper analysis and will create an effective system for identifying species in this group, leading to a major advance in our capacity to identify immature and male scale insects. Finally, this work has revealed that the barcode region of scale insects possesses both unusual nucleotide composition and a high incidence of indels, observations that suggest the value of studies to ascertain whole mitochondrial genomes in selected scale insects.
Acknowledgements
We thank Greg Evans (USDA, Animal and Plant Health Inspection, USA) for useful revisionary suggestions on an earlier draft of this manuscript. This research was supported by a grant (FDM0501011) from the National Plant Quarantine Service and by grants from Genome Canada through the Ontario Genomics Institute.
Supplementary material
The following supplementary material can be viewed online at http://journals.cambridge.org/ber.
S1. Information of samples used in this study.
S2. DNA sequences which have thymine residue deletions. This file was generated using MEGA software and hard copied to PowerPoint.
S3. The entire NJ tree of specimens generated using the ‘Sequence Analysis tool’ of BOLD.







