



Introduction
Definition of illness
Tourette syndrome (TS) is a childhood-onset chronic complex neuropsychiatric disorder characterized by multiple motor tics and at least one vocal tic persisting for more than 1 year (American Psychiatric Association, 2013; Robertson et al., Reference Robertson, Eapen, Singer, Martino, Scharf, Paschou and Leckman2017). Tics are defined by sudden, rapid, recurrent, nonrhythmic motor movements or vocalizations. They are generally involuntary and can include almost any muscle group or vocalization, although certain tics are common across TS patients, such as facial grimacing and eye blinking (American Psychiatric Association, 2013). Tics can be simple, involving only one group of muscles or sounds over a short duration, or complex, featuring coordinated patterns of movements, sounds, or speech that are longer in duration (American Psychiatric Association, 2013; Robertson et al., Reference Robertson, Eapen, Singer, Martino, Scharf, Paschou and Leckman2017). Tics wax and wane in severity, intensity and frequency, and they can persist, disappear and change in typology and topography over time (American Psychiatric Association, 2013; Robertson et al., Reference Robertson, Eapen, Singer, Martino, Scharf, Paschou and Leckman2017). Most individuals are able to suppress their tics, although only for a limited period of time and at the expense of unpleasantness (Leckman, Walker, & Cohen, Reference Leckman, Walker and Cohen1993). Although some studies have reported abnormalities in the cortico–striato–thalamo–cortical (CSTC) circuit of TS patients (Crittenden & Graybiel, Reference Crittenden and Graybiel2011; Singer, Reference Singer2013), the biology and molecular pathogenesis underlying this disorder remains unknown. Current treatments only provide partial relief of symptoms and often produce side effects (Robertson, Reference Robertson2000).
History
TS is named after Georges Albert Édouard Brutus Gilles de la Tourette, who in 1885 published the first clear description of this condition present in nine patients (Kushner, Reference Kushner2000; McNaught & Mink, Reference McNaught and Mink2011; Tourette, Reference Tourette1885). Although Gilles de la Tourette was the first person to formally describe symptoms, course of illness and predisposing cause of TS, earlier descriptions of tics exist. A document dated back to 1498 exists describing a priest who presented physical and vocal tics (McNaught & Mink, Reference McNaught and Mink2011). Patients with involuntary movements and vocalizations were reported in 1663 by William Drage (who reported the possible first case in UK, Mary Hall); 1825, by Jean Marc Gaspard Itard; 1873, by Armand Trousseau; and 1884, by Hughlings Jackson (McNaught & Mink, Reference McNaught and Mink2011; Robertson et al., Reference Robertson, Eapen, Singer, Martino, Scharf, Paschou and Leckman2017; 125 Years of Tourette Syndrome: The Discovery, Early History & Future of the Disorder, n.d.). By 1921, physicians such as Sandor Ferenczi began addressing TS from a psychoanalytical perspective (Ferenczi, Reference Ferenczi1921; Kushner, Reference Kushner2000; McNaught & Mink, Reference McNaught and Mink2011). It was not until the 1960–1970s that the psychoanalytical interpretation was questioned, when Arthur Shapiro began to treat TS patients with the neuroleptic drug haloperidol (McNaught & Mink, Reference McNaught and Mink2011; Shapiro & Shapiro, Reference Shapiro and Shapiro1968). When Shapiro and colleagues concluded that the etiology of Tourette symptoms was organic, and not a consequence of ‘bad parenting’, many more families began to connect by the early 1970s, establishing the US Tourette Syndrome Association (TSA) in 1972 (Kushner, Reference Kushner2000; Robertson et al., Reference Robertson, Eapen, Singer, Martino, Scharf, Paschou and Leckman2017). In 1980, TS was included in the third edition of the American Diagnostic and Statistical Manual of Mental Disorders (DSM-III), although tics had been included in the first edition of DSM, in 1952 (American Psychiatric Association, 1952; Robertson et al., Reference Robertson, Eapen, Singer, Martino, Scharf, Paschou and Leckman2017).
Diagnostic classification and criteria
In the DSM-5, TS belongs to a spectrum of neurodevelopmental conditions referred to as Tic Disorders, along with the persistent (chronic) motor or vocal tic disorder (which requires the presence of either motor or vocal tics), the provisional tic disorder (characterized by the presence of tics for less than a year), and the other specified and unspecified tic disorders (American Psychiatric Association, 2013).
The specific diagnostic criteria for TS in the latest edition of the DSM (DSM-5) are: (a) both multiple motor and one or more vocal tics have been present at some time during the illness, although not necessarily concurrently; (b) the tics may wax and wane in frequency but have persisted for more than 1 year since first tic onset; (c) onset is before age 18 years; and (d) the disturbance is not attributable to the physiological effects of a substance or another medical condition.
Different assessment tools have been developed in order to measure tic repertoire and severity in an individual. Currently, the most commonly used is the Yale Global Tic Severity Scale, which provides an evaluation of the number, frequency, intensity, complexity, and interference of motor and phonic symptoms (Leckman et al., Reference Leckman, Riddle, Hardin, Ort, Swartz, Stevenson and Cohen1989).
Epidemiology (lifetime prevalence estimates, risk factors, morbidity, mortality, and comorbidities)
Prevalence, morbidity, comorbidities, and mortality
TS has a worldwide estimated prevalence of 0.3–1% in children and can lead to a substantial decrease in quality of life, especially when tics are severe or when other comorbidities are present (American Psychiatric Association, 2013; Knight et al., Reference Knight, Steeves, Day, Lowerison, Jette and Pringsheim2012; Robertson, Reference Robertson2008; Scharf et al., Reference Scharf, Miller, Gauvin, Alabiso, Mathews and Ben-Shlomo2015). Notably, there is a well-established gender imbalance in the prevalence of TS, with a male-to-female ratio of 2–4:1 (American Psychiatric Association, 2013; Freeman et al., Reference Freeman, Fast, Burd, Kerbeshian, Robertson and Sandor2000; Knight et al., Reference Knight, Steeves, Day, Lowerison, Jette and Pringsheim2012). Tics present at an average age at onset of 4–6 years, typically reach the peak severity between 10 and 12 years, and decrease in severity during adolescence for 50–75% of patients (American Psychiatric Association, 2013; Leckman et al., Reference Leckman, Zhang, Vitale, Lahnin, Lynch, Bondi and Peterson1998). Groth et al. published a large prospective clinical study where they indicate that, in adulthood, most cases (59.5%) have a mild form of the disorder, with remission reported in 17.7% of diagnoses (Groth, Mol Debes, Rask, Lange, & Skov, Reference Groth, Mol Debes, Rask, Lange and Skov2017). A small group (22.8%) presented moderate or severe and debilitating tic symptoms (Groth et al., Reference Groth, Mol Debes, Rask, Lange and Skov2017). Adult TS is notable for being associated with a lifelong disability in 15% of cases (Elstner, Selai, Trimble, & Robertson, Reference Elstner, Selai, Trimble and Robertson2001; Leckman, Bloch, Scahill, & King, Reference Leckman, Bloch, Scahill and King2006).
More than 85% of individuals with TS present a lifetime diagnosis of any neuropsychiatric disorders (Hirschtritt et al., Reference Hirschtritt, Lee, Pauls, Dion, Grados and Illmann2015). Among the most common comorbid disorders are obsessive-compulsive disorder (OCD, 50%) and attention deficit hyperactivity disorder (ADHD, 54%) (Hirschtritt et al., Reference Hirschtritt, Lee, Pauls, Dion, Grados and Illmann2015). Other conditions have also been described in patients with TS including major depression, non-OCD anxiety, conduct disorder, learning disorder, as well as mood and disruptive behavior (Hirschtritt et al., Reference Hirschtritt, Lee, Pauls, Dion, Grados and Illmann2015).
In some instances, TS includes physical, orthopedic or neurological self-injury related to forceful movements (American Psychiatric Association, 2013; Stafford & Cavanna, Reference Stafford and Cavanna2020), and when compared to controls, TS/chronic tic disorder (CTD) patients have been shown to have higher risk for attempting suicide or dying by suicide (Fernández de la Cruz et al., Reference Fernández de la Cruz, Rydell, Runeson, Brander, Rück, D'Onofrio and Mataix-Cols2017). In a nationwide prospective study comprising children born between 1960 and 2012 in Denmark, the all-cause mortality rate ratio (including suicide causes) has been reported to be 1.63 in TS patients compared to controls, and 1.39 after adjusting for comorbidities (Meier, Dalsgaard, Mortensen, Leckman, & Plessen, Reference Meier, Dalsgaard, Mortensen, Leckman and Plessen2017).
Etiology
Analyses of TS heritability suggest a complex etiology that includes multiple risk factors. The most well-evidenced set of risk factors are those that are genetic. Much of the progress in our study of the genetic risk factors has only been made in the last few decades (Fig. 1).
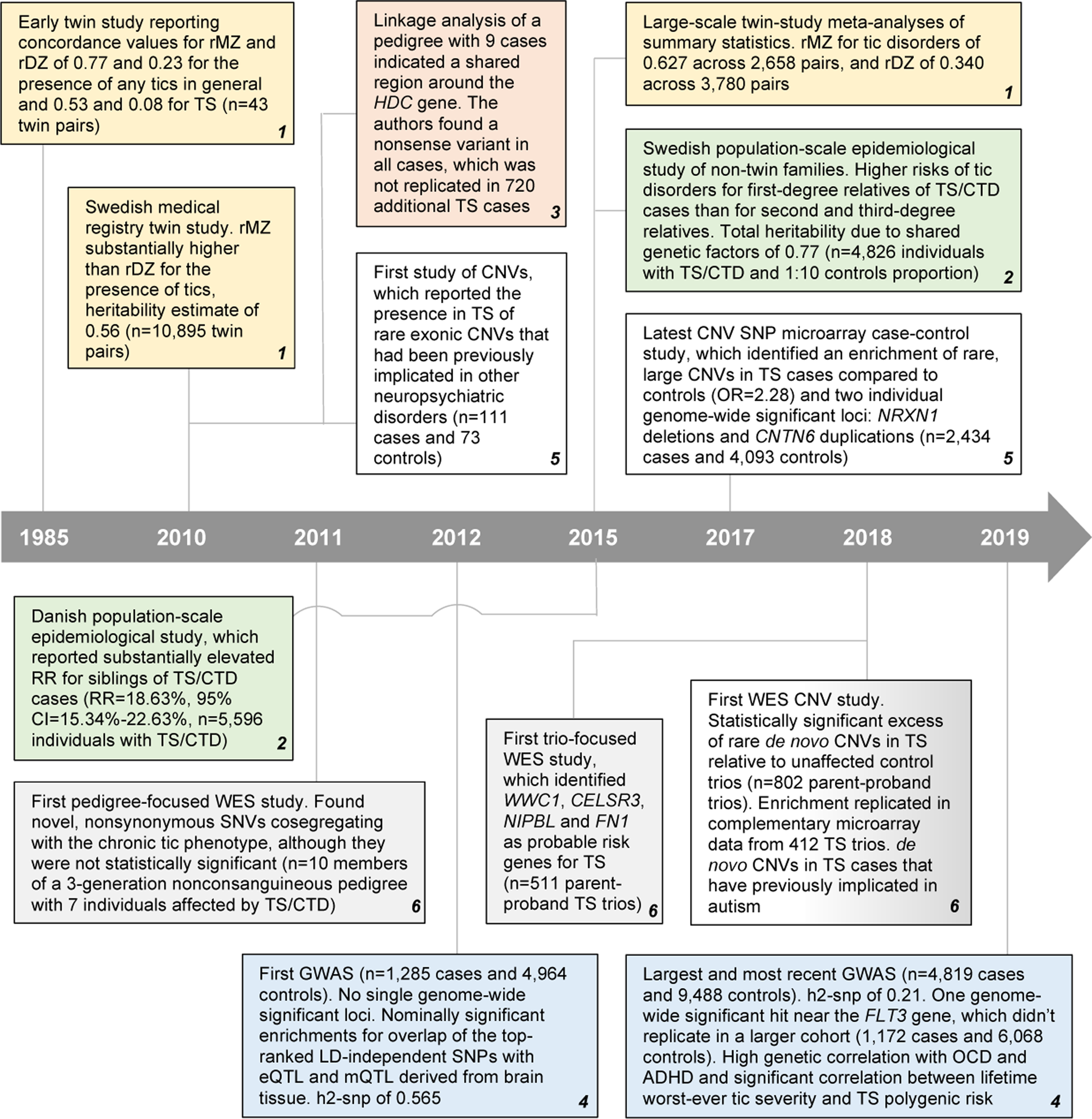
Fig. 1. Key genetic studies and findings in TS. Adapted from Robertson et al. (Reference Robertson, Eapen, Singer, Martino, Scharf, Paschou and Leckman2017). Twin studies in yellow; non-twin studies in green; linkage studies in salmon; genome-wide association studies (GWAS) in blue; rare copy number variant (CNVs) in white; whole-exome sequencing (WES) in gray. TS, Tourette syndrome; OCD, obsessive-compulsive disorder; ADHD, attention deficit hyperactivity disorder; CTD, chronic tic disorder; rMZ, monozygotic twin correlation (for diagnosis); rDZ, dizygotic twin correlation (for diagnosis); SNP, single-nucleotide polymorphism; LD, linkage disequilibrium; eQTL, expression quantitative trait locus; mQTL, methylation quantitative trait locus; h2-snp, SNP-based heritability estimate; RR, relative risk; HDC, histidine decarboxylase; NRXN1, neurexin 1.
Apart from TS risk factors, there are additional factors that can increase tic severity. According to the DSM-5, they can be divided into three different groups: (a) temperamental (anxiety, stress, excitement, and exhaustion); (b) environmental (e.g. observing gestures or sounds in other persons may result in echopraxia or echolalia, high temperatures); and (c) genetic and physiological (genetic variation, obstetrical complications, older paternal age, lower birth weight, and maternal smoking during pregnancy) (American Psychiatric Association, 2013; Robertson et al., Reference Robertson, Eapen, Singer, Martino, Scharf, Paschou and Leckman2017).
Genetic epidemiology
Twin studies
Twin studies focused on TS and the more generalized presence of tics have offered longstanding support for a genetic contribution to risk. An early study reported diagnostic concordance values for mono- and di-zygotic twins (rMZ and rDZ, respectively) of 0.77 and 0.23 for the presence of any tics in general, and a lower rMZ and rDZ of 0.53 and 0.08 for formal TS diagnosis (Price, Kidd, Cohen, Pauls, & Leckman, Reference Price, Kidd, Cohen, Pauls and Leckman1985). In spite of this study only featuring 43 twin pairs and thus being underpowered, it suggested that tic severity itself is a complex trait with genetic and non-genetic contributions, and that it is more likely for monozygotic twins to both have the presence of tics than it for them to both be diagnosed with full-blown TS. An expansive twin study utilizing the Swedish medical registry (n = 10 895 twin pairs) noted rMZ and rDZ values in line with initial estimates, and an rMZ substantially higher than rDZ (Lichtenstein, Carlström, Råstam, Gillberg, & Anckarsäter, Reference Lichtenstein, Carlström, Råstam, Gillberg and Anckarsäter2010). It also reported a twin-based heritability estimate (i.e. concordance of presence/absence of the trait across twin pairs) of 0.56 for the presence of tics (Lichtenstein et al., Reference Lichtenstein, Carlström, Råstam, Gillberg and Anckarsäter2010). Most recently, large-scale meta-analyses of summary statistics reported for 17 804 traits across 2748 publications led to an rMZ for Tic Disorders of 0.63 across 2658 pairs, and an rDZ of 0.34 across 3780 pairs, in line with initial estimates (Polderman et al., Reference Polderman, Benyamin, de Leeuw, Sullivan, van Bochoven, Visscher and Posthuma2015). These concordance rates are lower than other more well-studied neuropsychiatric disorders such as schizophrenia and bipolar disorder (rMZ of 0.76 and 0.82 across 19 841 and 9251 pairs respectively) (Polderman et al., Reference Polderman, Benyamin, de Leeuw, Sullivan, van Bochoven, Visscher and Posthuma2015), suggesting a greater comparative role for the influence of non-genetic factors on TS risk.
Non-twin family studies
Population-scale epidemiological studies of non-twin families also support genetic contribution to tics (Browne et al., Reference Browne, Hansen, Buxbaum, Gair, Nissen, Nikolajsen and Grice2015; Mataix-Cols et al., Reference Mataix-Cols, Isomura, Pérez-Vigil, Chang, Rück, Larsson and Lichtenstein2015). Research of thorough, population-wide registry data from Scandinavia has proven the most appealing subject of these studies. One key example is a study focused on the Swedish registry to identify 4826 individuals with TS or CTD, and matched them at a 1:10 ratio with unaffected, unrelated individuals from the registry (Mataix-Cols et al., Reference Mataix-Cols, Isomura, Pérez-Vigil, Chang, Rück, Larsson and Lichtenstein2015). Researchers then compared the likelihood of affected individuals having a first-degree relative with TS or CTD relative to matched unaffected individuals. Consistent with rDZ twin concordance estimates, first-degree relatives of TS/CTD cases were far more likely to have TS/CTD themselves [odds ratio (OR) 18.69, 95% confidence interval (CI) 14.53–24.05], as were second-degree relatives (OR 4.58, 95% CI 3.22–6.52) and third-degree relatives (OR 3.07, 95% CI 2.08–4.51). Such substantial increases in relative risk are consistent with a subset of risk factors that are genetic in origin. This study estimated the total heritability due to shared genetic factors at 0.77, with the remainder being attributed to non-shared environmental effects. Although earlier twin studies reported a higher amount of genetic heritability between male twins (Qi, Zheng, Li, & Xiong, Reference Qi, Zheng, Li and Xiong2017), this study failed to find additional evidence in support of this, instead suggesting tic heritability levels in males and females are similar. At around the same time, a similar study was published focused specifically on Danish registry data (Browne et al., Reference Browne, Hansen, Buxbaum, Gair, Nissen, Nikolajsen and Grice2015). Out of around 1.7 million included Danish individuals born from 1980 to 2007, it identified 5596 individuals total with a diagnosis of TS or CTD that were suitable for analysis. It reported a substantially elevated recurrence risk for siblings of TS/CTD cases (18.63%, 95% CI 15.34–22.63) (Browne et al., Reference Browne, Hansen, Buxbaum, Gair, Nissen, Nikolajsen and Grice2015), in line with the elevated risk observed in first-degree relatives of cases in the Swedish study. Both studies offer strong lines of evidence that within large populations, TS/CTD cluster within families (Browne et al., Reference Browne, Hansen, Buxbaum, Gair, Nissen, Nikolajsen and Grice2015; Mataix-Cols et al., Reference Mataix-Cols, Isomura, Pérez-Vigil, Chang, Rück, Larsson and Lichtenstein2015). The most plausible explanation for this clustering is a genetic contribution to TS/CTD risk.
Genetics
Linkage studies
Linkage-based approaches have thus far not yielded a substantial number of findings in published research in TS. Initial studies focused on large, multi-generational pedigrees operated on the theory that the presence of multiple TS-affected individuals within these pedigrees were due to the presence of one or a few highly penetrant genetic risk factors (Curtis, Robertson, & Gurling, Reference Curtis, Robertson and Gurling1992; Eapen, Pauls, & Robertson, Reference Eapen, Pauls and Robertson1993; Kurlan et al., Reference Kurlan, Behr, Medved, Shoulson, Pauls, Kidd and Kidd1986). Most studies simply describe the results of linkage study, where segments that are shared to a nominal degree of significance are described. Although linkage peaks have been observed in individual family structures, none replicate across families, and none (save for one, that has never been replicated) have met strict statistical criteria for being a genome-wide significant linkage peak (O'Rourke, Scharf, Yu, & Pauls, Reference O'Rourke, Scharf, Yu and Pauls2009). One example in line with the autosomal dominant model initially proposed with pedigrees such as these is (Ercan-Sencicek et al., Reference Ercan-Sencicek, Stillman, Ghosh, Bilguvar, O'Roak, Mason and State2010), where a two-generation non-consanguineous family is described consisting of an affected father, unaffected mother and eight children, all of which are affected. Linkage analysis indicated a shared region in a 3.4 cM region of chromosome 15, around the histidine decarboxylase (HDC) gene region. Subsequently, researchers found that the father was heterozygous for a stop-gain variant within this gene that had been transmitted to all eight affected children. Researchers failed to find additional HDC stop-gain variants in an additional 720 TS cases. Thus far, de novo coding HDC mutations in TS have not been observed in more recent studies (Wang et al., Reference Wang, Mandell, Kumar, Sun, Morris, Arbelaez and State2018). One follow-up hypothesis that is insufficiently explored in earlier pedigree study but now being explored more fully in pedigree studies of psychiatric cohorts is that families with a large number of affecteds may carry a heavy load of common risk variation (de Jong et al., Reference de Jong, Diniz, Saloma, Gadelha, Santoro, Ota and Breen2018; Szatkiewicz et al., Reference Szatkiewicz, Crowley, Adolfsson, Åberg, Alaerts, Genovese and Sullivan2019). It is also possible that, in some of these pedigrees, higher-penetrance rare noncoding risk variants are present in the data that have not been detected via current selection criteria that are biased heavily toward coding annotations. As our understanding of the noncoding genome accelerates, our ability to identify the subset of these variants that are more likely to make substantial contributions to risk will increase.
Genome-wide association studies of common variation with TS
The first TS genome-wide association study (GWAS) of common genetic variants (allele frequency ⩾0.01) included 1285 cases and 4964 controls, and was underpowered for common risk variant detection (Scharf et al., Reference Scharf, Yu, Mathews, Neale, Stewart, Fagerness and Pauls2013). No single genome-wide significant loci were detected in the study, but analysis of the summary statistics suggested that this was due to insufficient sample size. In assessing the top-ranked linkage disequilibrium (LD)-independent single nucleotide polymorphisms (SNPs) (412 with p < 0.001) nominally significant enrichments were noted for overlap with expression and methylation quantitative trait loci (eQTL and mQTL respectively) derived from brain tissue.
The largest and most recent TS GWAS included newly genotyped cases and controls (referred to as GWAS2) alongside samples from the prior described set of results (herein referred to as GWAS1) for a combined analysis consisting of 4819 TS cases and 9488 controls (Yu et al., Reference Yu, Sul, Tsetsos, Nawaz, Huang and Zelaya2019). It reports a trait heritability derived from common genetic variation (h2-snp) across the full cohort of 0.21 with a standard error (s.e.) of 0.024. Authors note that the GWAS1 cohort h2-snp (0.57, s.e. = 0.1, p = 1 × 10−9) is substantially higher than that of the newest ‘GWAS2’ batch of samples (0.29, s.e. = 0.04, p = 6 × 10−14). It is reasoned that this is due to cases within GWAS1 being enriched for the presence of at least one affected sibling (i.e. multiplex family history), which would be consistent with a particularly heavy load of common risk variation within these samples. Across the full meta-analysis, one genome-wide significant hit was detected, near the Fms Related Receptor Tyrosine Kinase 3 (FLT3) gene. The locus failed to replicate in an independent cohort of 1172 cases and 6068 controls, a standard in statistical genetics that is generally required to be met in order to formally implicate an individual variant with risk.
Authors of the most recent TS GWAS also found that the GWAS summary statistics could be used to calculate sample-level genetic risk Scores (GRS) that were predictive of TS case status and of tic severity (Yu et al., Reference Yu, Sul, Tsetsos, Nawaz, Huang and Zelaya2019). GRS calculation in human samples was adapted from agricultural genetic work, where it had been used to more effectively select crops and livestock for breeding that carry preferable traits. Although contemporary methods have grown more complex, GRS values are in general calculated for single samples by taking independent variation associated with a trait at some level of significance (say, p < 0.05) and defining the risk score as a weighted sum of the number of alleles for each variant multiplied by the relative risk or protection conferred by each allele copy. In doing this with the TS summary statistics, authors found that TS GRS values calculated across an independent population-based sample was predictive of TS case status (OR 1.33, p = 5 × 10−9) and of tic spectrum case status (OR 1.20, p = 5 × 10−4). In a manner consistent with heritability calculations in multiplex-dominated GWAS1 v. GWAS2, authors also reported that multiplex TS cases carried higher GRS values than simplex TS cases (p = 0.027). Finally, they noted a significant positive correlation between case GRS values and recorded worst-ever tic severity (β = 0.93, s.e. = 0.42, p = 0.03). This correlation is consistent with the spectrum nature of tic severity and tic diagnoses, and suggests that individuals with more severe CTD and formal TS are more likely to carry high genetic burden for TS.
Critically, data from the most recent TS GWAS provides genetic insight into the comorbidities observed for the disorder. We computed genetic correlations between the TS dataset and publicly available GWAS datasets for other psychiatric disorders using LDSC v1.0.1 (Bulik-Sullivan et al., Reference Bulik-Sullivan, Loh, Finucane, Ripke, Yang and Neale2015). The correlation patterns were concordant with comorbidities observed in TS patients (Fig. 2). Specifically, a particularly high genetic correlation with OCD is observed (r g = 0.42, s.e. = 0.09), along with a high genetic correlation with ADHD (r g = 0.21, s.e. = 0.05). As described earlier, these disorders are the most commonly described TS comorbidities. Interestingly, although anorexia nervosa (AN) and OCD are highly comorbid with one another, and have been reported to have a high genetic correlation (Yilmaz et al., Reference Yilmaz, Halvorsen, Bryois, Yu, Thornton and Zerwas2018), the genetic correlation between TS and AN is very low. This result is in line with TS comorbidity statistics, and suggests that genetic architecture shared between TS and OCD is distinct from the architecture shared between OCD and AN.
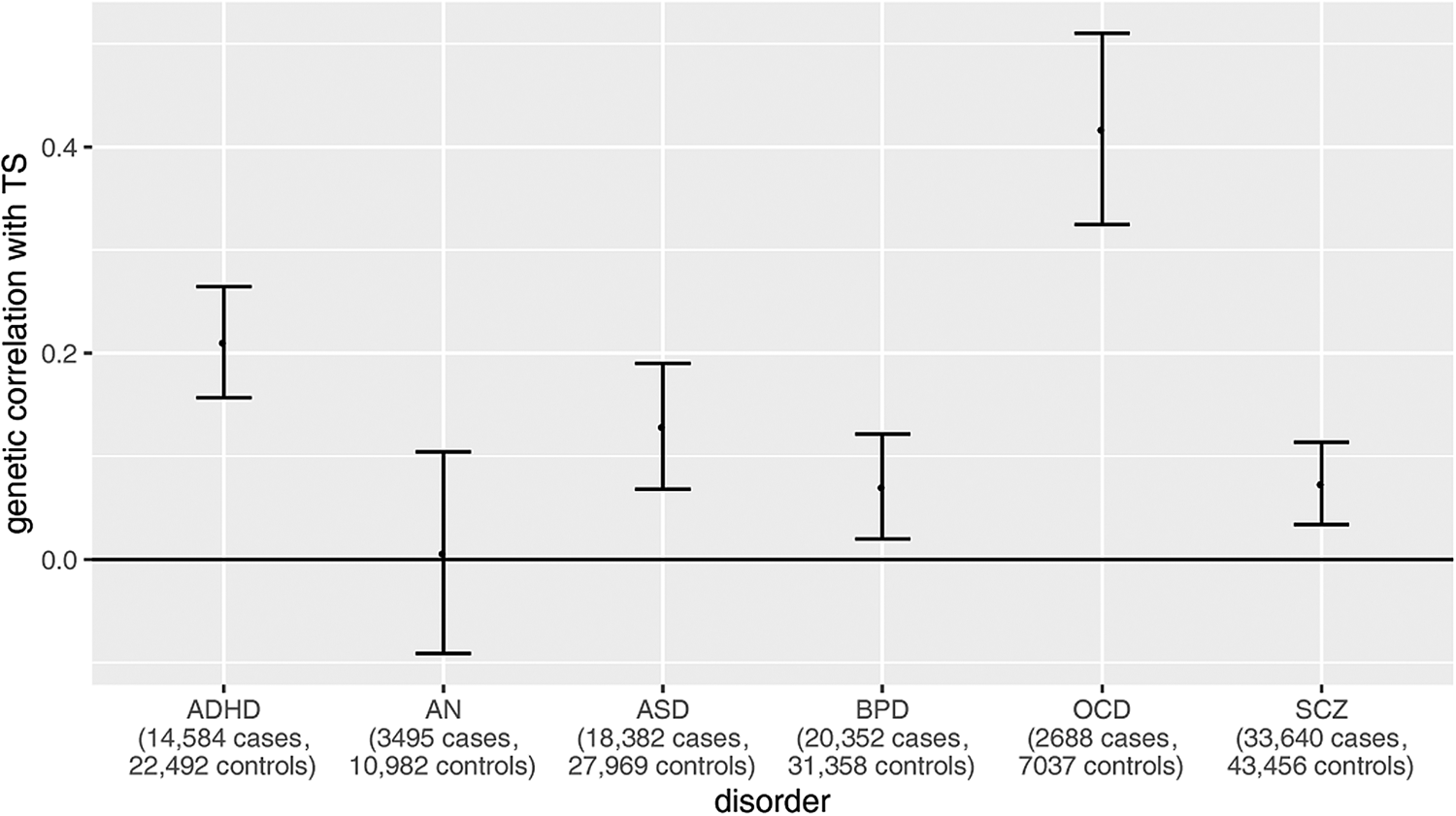
Fig. 2. Genetic correlations between TS and other neuropsychiatric disorders from available GWAS summary statistics. Publicly available GWAS statistics for TS (4819 cases, 9488 controls) and for six separate neuropsychiatric disorders [ADHD (Demontis et al., Reference Demontis, Walters, Martin, Mattheisen, Als, Agerbo and Neale2019); AN (Duncan et al., Reference Duncan, Yilmaz, Gaspar, Walters, Goldstein, Anttila and Bulik2017); ASD (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019); BD (Stahl et al., Reference Stahl, Breen, Forstner, McQuillin, Ripke and Trubetskoy2019); OCD (International Obsessive Compulsive Disorder Foundation Genetics Collaborative (IOCDF-GC) and OCD Collaborative Genetics Association Studies (OCGAS), 2018); SCZ (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014)] were obtained for genetic correlation calculations using LDSC v1.0.1 (Bulik-Sullivan et al., Reference Bulik-Sullivan, Loh, Finucane, Ripke, Yang and Neale2015). Sample sizes have been included for each disorder on the X axis. Significant genetic correlations are defined as those with a p value reported by LDSC less than the Bonferroni threshold, here defined as 0.05/6 tests = 8 × 10−3. Only ADHD and OCD have genetic correlations with TS that pass this threshold. These disorders have been noted as among the highest comorbid diagnoses in TS cases.
Rare copy-number variants
Rare copy-number variants (CNVs) have been shown to be significant risk factors for several neuropsychiatric disorders (Levy, Xu, Gogos, & Karayiorgou, Reference Levy, Xu, Gogos and Karayiorgou2012), including for TS (Levy et al., Reference Levy, Xu, Gogos and Karayiorgou2012; McGrath et al., Reference McGrath, Yu, Marshall, Davis, Thiruvahindrapuram, Li and Scharf2014; Nag et al., Reference Nag, Bochukova, Kremeyer, Campbell, Muller, Valencia-Duarte and Ruiz-Linares2013; Sundaram, Huq, Wilson, & Chugani, Reference Sundaram, Huq, Wilson and Chugani2010). CNVs involve the deletion or duplication of large genomic regions (often over 100 000 base pairs) (1000 Genomes Project Consortium et al., Reference Auton, Brooks, Durbin, Garrison, Kang and Abecasis2015; Mills et al., Reference Mills, Walter, Stewart, Handsaker, Chen and Alkan2011; Redon et al., Reference Redon, Ishikawa, Fitch, Feuk, Perry, Andrews and Hurles2006; Sudmant et al., Reference Sudmant, Rausch, Gardner, Handsaker, Abyzov, Huddleston and Korbel2015) and as such, particularly large CNVs that impact the function of multiple genes have been implicated with neuropsychiatric disorders (Guyatt et al., Reference Guyatt, Stergiakouli, Martin, Walters, O'Donovan, Owen and Gaunt2018; Levy et al., Reference Levy, Xu, Gogos and Karayiorgou2012; Martin et al., Reference Martin, Tammimies, Karlsson, Lu, Larsson, Lichtenstein and Magnusson2019). The first CNV-focused study of TS was performed on DNA microarray data from 111 patients and 73 controls and indicated the presence of rare exonic CNVs in TS that had been previously implicated in other neuropsychiatric disorders, such as neurexin 1 (NRXN1) and catenin, alpha3 (CTNNA3) (Sundaram et al., Reference Sundaram, Huq, Wilson and Chugani2010). These results suggested an overlap between CNV risk architecture for TS and for the other more well-studied neuropsychiatric disorders. Four other studies also found an overlap of rare CNVs between TS and autism spectrum disorder (ASD) (Huang et al., Reference Huang, Yu, Davis, Sul, Tsetsos and Ramensky2017; Levy et al., Reference Levy, Xu, Gogos and Karayiorgou2012; McGrath et al., Reference McGrath, Yu, Marshall, Davis, Thiruvahindrapuram, Li and Scharf2014; Nag et al., Reference Nag, Bochukova, Kremeyer, Campbell, Muller, Valencia-Duarte and Ruiz-Linares2013). The study from Fernandez et al. included 460 TS cases and 1131 controls, and although they didn't find a significant increase in the number of rare CNVs in cases v. controls, they detected rare CNVs in TS overlapping genes previously identified in ASD (Fernandez et al., Reference Fernandez, Sanders, Yurkiewicz, Ercan-Sencicek, Kim, Fishman and State2012). Moreover, they noted an enrichment of CNVs overlapping genes involved in in histaminergic and GABAergic pathways in TS cases. Another study performed in two Latin American populations, which included 179 TS cases and 234 controls, identified two cases with ~400 kb deletions overlapping NRXN1 and four duplications in Collagen Type VIII Alpha 1 Chain (COL8A1) (Nag et al., Reference Nag, Bochukova, Kremeyer, Campbell, Muller, Valencia-Duarte and Ruiz-Linares2013). They validated these results identifying additional patients with rearrangements in NRXN1 and COL8A1, which were not found in controls. They also found that cases had a significant excess of large CNVs (>500 kb) compared to controls. In 2014, McGrath et al. performed a cross-disorder study of CNVs in TS and OCD, involving 1086 TS, 1613 OCD, and 1789 control samples (McGrath et al., Reference McGrath, Yu, Marshall, Davis, Thiruvahindrapuram, Li and Scharf2014). Although they didn't find a global CNV burden in the cross-disorder study or in the separate secondary analyses, they found a 3.3-fold increased burden of large deletions previously associated with other neurodevelopmental disorders in patients with TS/OCD.
The latest study using SNP microarray data analyzed 2434 TS cases and 4093 controls for rare CNVs and identified an excess of rare, large CNVs in TS cases compared to controls (OR 2.28, 95% CI 1.39–3.79, p = 1.2 × 10−3) (Huang et al., Reference Huang, Yu, Davis, Sul, Tsetsos and Ramensky2017). Moreover, they also identified two types of CNVs associated with TS risk at genome-wide significance: NRXN1 deletions (OR 20.3, 95% CI 2.6–156.2) and CNTN6 duplications (OR 10.1, 95% CI 2.3–45.4). More recently, Wang et al. analyzed 802 TS parent-proband trios from whole-exome sequencing (WES) and showed a statistically significant excess of rare de novo CNVs in TS relative to unaffected control trios (rate ratio = 2.2; p = 2.5 × 10−3) (Wang et al., Reference Wang, Mandell, Kumar, Sun, Morris, Arbelaez and State2018). In addition, they replicated this enrichment in complementary microarray data from 412 TS trios (rate ratio = 2.8; p = 0.024) and found de novo CNVs in TS cases that have been previously implicated with ASD (Sanders et al., Reference Sanders, He, Willsey, Ercan-Sencicek, Samocha, Cicek and State2015). These results in full suggest that the study of CNVs in larger cohorts may identify additional rare CNVs conferring risk for TS.
WES and its initial application to pedigrees
High throughput sequencing has allowed for the expansion of the focus of rare variant studies beyond CNVs to include insertion–deletions (indels) and single-nucleotide variants (SNVs). An effective implementation of this technology has been WES, where the ~1% of the human genome that codes for protein is specifically targeted for sequencing and variant discovery. Within psychiatric genetics this sequencing approach has been used to detect rare coding variants that are near-absent from the general population, some of which are inherited from parents while others are absent from both parents (i.e. de novo in origin). Genetic studies focused on these variants have identified single high-confidence risk genes and have helped to elucidate the biology underlying psychiatric disease.
WES was first applied to TS in a pedigree-focused context. The first study that applied WES to TS was focused on the role of rare coding variants inherited in multiplex families. WES was conducted on DNA from 10 members of a three-generation nonconsanguineous pedigree with seven individuals affected by TS/CTD, and found novel, nonsynonymous SNVs co-segregating with the chronic tic phenotype in three separate genes – Mitochondrial Ribosomal Protein L3 (MRPL3), DnaJ Heat Shock Protein Family (Hsp40) Member C13 (DNAJC13), and Orofacial Cleft 1 Candidate 1 (OFCC1) (Sundaram et al., Reference Sundaram, Huq, Sun, Yu, Bennett, Wilson and Chugani2011). In 2018, another group applied WES to DNA from six members of a three-generation nonconsanguineous TS multiplex family, and found a rare heterozygous nonsense mutation in PNKD Metallo-Beta-Lactamase Domain Containing (PNKD) that co-segregated with TS. The group characterized the functional consequences of the mutation in iPSC-derived neurons and demonstrated that the mutation was associated with the haploinsufficiency of some isoforms of the gene (Sun et al., Reference Sun, Nasello, Deng, Wang, Zhang, Xu and Tischfield2018). The findings in these pedigrees were not subjected to a specific test of statistical significance, and the individual pedigree did not have genome-wide significance for linkage. Thus, all of these genes require additional independent evidence before being classified as TS risk genes.
De novo coding SNVs and indels
Three studies so far have applied WES to investigate rare de novo variants in large TS simplex cohorts. Willsey et al. called de novo coding variants across 511 TS trios and using a Transmission and De novo Association (TADA) framework, identified WW and C2 domain containing 1 (WWC1), cadherin EGF LAG seven-pass G-type receptor 3 (CELSR3), NIPBL cohesin loading factor (NIPBL), and fibronectin 1 (FN1) as probable risk genes for TS (Willsey et al., Reference Willsey, Fernandez, Yu, King, Dietrich, Xing and Heiman2017). The sequencing of 291 additional TS trios led to a follow-up analysis on a total of 802 trios by Wang and colleagues, who identified CELSR3 as a high-confidence TS risk gene and observed an enrichment of de novo damaging variants in genes involved in cell polarity (Wang et al., Reference Wang, Mandell, Kumar, Sun, Morris, Arbelaez and State2018). The study also found a statistically significant excess of damaging de novo variants in mutation-intolerant genes in simplex families but not in multiplex families. Across included trios, around 10% of clinical cases carried de novo damaging coding variants that likely mediate TS risk, and authors estimated that around 483 genes total may contribute to TS risk when impacted by these variants. Additionally, they noted an overlap of de novo sequence variants between TS and OCD, consistent with shared genetic risk. Liu et al. (Reference Liu, Tian, He, Li, Xie, Liu and Guan2020) analyzed WES data from 97 TS trios and identified a de novo missense variant in the neurodevelopmental risk gene ASH1L alongside some evidence of missense variant over-transmission from parents to probands in the cohort (Coe et al., Reference Coe, Stessman, Sulovari, Geisheker, Bakken, Lake and Eichler2019; Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Buxbaum2020). Finally, de novo variants from a much smaller cohort of 15 trios were described in a fourth smaller study from Zhao et al. (Reference Zhao, Wang, Hao, Zhu, Zhang and Wu2020), including a missense variant in the neurodevelopmental gene KMT2C (Coe et al., Reference Coe, Stessman, Sulovari, Geisheker, Bakken, Lake and Eichler2019; Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Buxbaum2020).
Potential for whole genome sequencing studies of TS
Whole genome sequencing (WGS) could be applied to TS cases to fully characterize genetic contributions to overall risk. WGS allows for the complete characterization of common and rare variation present in each patient. These include all variants detectable in array and WES data, along with rare noncoding SNVs and indels that are outside the reach of these other assays. WGS can also be used to detect CNV deletions and duplications that are too small to be reliably detected in array data, along with inversions and translocations that are undetectable via array or WES (Mills et al., Reference Mills, Walter, Stewart, Handsaker, Chen and Alkan2011). Analyses of WGS data have proven difficult, in part because deleterious noncoding variants are still not well-defined (Halvorsen et al., Reference Halvorsen, Huh, Oskolkov, Wen, Netotea, Giusti-Rodriguez and Szatkiewicz2020). In addition, the computational burden of storage and analysis is higher for WGS than for array or WES. With costs of WGS decreasing over time, it is too early to make a final judgment on whether or not WGS yields good returns on investment for the study of psychiatric disease.
Study of postmortem TS brain tissue
Existing studies focused on postmortem TS brain tissue are small in number and in sample size. Neuroimaging studies have consistently reported abnormalities of the CSTC circuit function in TS pathophysiology (Albin & Mink, Reference Albin and Mink2006; Felling & Singer, Reference Felling and Singer2011). TS patients have been observed to have lower volumes of basal ganglia, a brain region which is connected to the CSTC and exerts control over motor function (Felling & Singer, Reference Felling and Singer2011; Peterson et al., Reference Peterson, Thomas, Kane, Scahill, Zhang, Bronen and Staib2003). Two studies focused on postmortem brain tissue from a single set of five TS cases have investigated alterations in neuronal cell populations. Both studies report a decrease in GABAergic and cholinergic interneurons in the striatum, a component of the basal ganglia (Kalanithi et al., Reference Kalanithi, Zheng, Kataoka, DiFiglia, Grantz, Saper and Vaccarino2005; Kataoka et al., Reference Kataoka, Kalanithi, Grantz, Schwartz, Saper, Leckman and Vaccarino2010). Aligning with this, a transcriptome study of postmortem brain tissue from nine TS cases v. nine matched controls describes lower case expression of transcripts preferentially expressed in striatal interneurons (Lennington et al., Reference Lennington, Coppola, Kataoka-Sasaki, Fernandez, Palejev, Li and Vaccarino2016).
Discussion
Current literature support a significant role for genetics in TS etiology. The concordance of TS and more generalized presence of tics in twin studies are consistent with a genetic contribution to risk. Studies of medical registry data report that having relatives with TS/CTD is a significant risk factor for TS/CTD diagnosis. Along the same lines, twin studies indicate that the heritability of tic disorders is higher when a broad phenotype is used, rather than TS strictly defined (Price et al., Reference Price, Kidd, Cohen, Pauls and Leckman1985). These findings imply that at the genetic level, TS/CTD fall along a continuum and not into the distinct categories implied by the current diagnostic criteria (Insel et al., Reference Insel, Cuthbert, Garvey, Heinssen, Pine, Quinn and Wang2010). Although early studies of individual large pedigrees did not yield strongly-evidenced findings that were easily-replicated, subsequent studies across thousands of cases have found evidence for a genetic architecture that is specific to TS risk.
Current GWAS focused on TS support a significant contribution from common genetic variation to overall TS risk. Two TS-focused GWAS have been published, each with no genome-wide significant hits that have survived replication analyses. In the most recent analysis, a significant overall SNP-based heritability was noted across the cohort, with a higher heritability estimate obtained from the subset of the cohort that came from multiplex family structures. Individual-level GRS values calculated across samples in these data were associated with both TS and the presence of tics, along with lifetime worst-ever tic severity scores (Yu et al., Reference Yu, Sul, Tsetsos, Nawaz, Huang and Zelaya2019). These findings are consistent with TS genetic risk from common variation acting in a dosage dependent manner, with higher dosing of the risk alleles leading to greater tic severity and a higher likelihood of TS diagnosis.
Studies of rare variation have proven effective at identifying specific risk genes for TS. So far, two genic rare CNVs (deletions overlapping NRXN1 and duplications overlapping CNTN6) have been identified as significant genetic risk factors for TS risk (Huang et al., Reference Huang, Yu, Davis, Sul, Tsetsos and Ramensky2017). In addition, Wang and colleagues report that de novo genic CNVs are associated with TS, particularly within simplex families (without a family history of a tic disorder) (Wang et al., Reference Wang, Mandell, Kumar, Sun, Morris, Arbelaez and State2018). In studies of de novo coding SNVs and indels, a total of six genes that are at least classifiable as probable risk genes have been identified thus far, including CELSR3 which is classifiable as a high confidence TS risk gene (Wang et al., Reference Wang, Mandell, Kumar, Sun, Morris, Arbelaez and State2018). Such genes likely represent core risk genes whose perturbation is directly related to TS etiology. As with other similar studies on different psychiatric disorders, future studies will need larger sample sizes in order to identify additional core risk genes for TS (Iakoucheva, Muotri, & Sebat, Reference Iakoucheva, Muotri and Sebat2019).
For the risk genes identified so far (n = 8 with status of at least ‘probable’ or declared as genome-wide significant) it is likely that the function of all is directly relevant to TS neurobiology and pathogenesis. Wang et al. noted that two of their six identified genes (CELSR3 and WWC1) have roles in establishing cell polarity, and found that genes with this precise role were enriched in damaging mutations (Wang et al., Reference Wang, Mandell, Kumar, Sun, Morris, Arbelaez and State2018). Ultimately, it is likely that when perturbed, TS risk genes contribute to the development of the disorder through a variety of effects on neurodevelopment and brain functionality, and that the disruption of this process is simply one of many involved in TS etiology.
Significant progress has been made in deducing and quantifying the genetic contribution of common and rare variation to TS risk. Through collaborative study in consortia that include the Tourette Syndrome Association International Consortium for Genetics (TSAICG) and Tourette International Collaborative (TIC) Genetics in the United States, and the European Multicentre Tics in Children Studies (EMTics) in the European Union, researchers are actively working to identify additional common and rare risk variation, and the TS risk genes that are impacted by them.
Acknowledgements
We are grateful to Drs. Jeremiah Scharf and Manuel Mattheisen for their valuable feedback on the review content. We thank the Psychiatric Genomics Consortium and its membership for providing an inclusive, welcoming community of scientists and physicians that have facilitated the process of writing this review.
Financial support
We received no specific grant from any funding agency, commercial, or not-for-profit sectors for writing this review.
Conflict of interest
The authors have no conflicts of interest to declare.


