Introduction
Ring chromosome 21 syndrome [r(21)] is a rare condition that was firstly described by Lejeune et al. in 1964 Reference Lejeune(1) and may occur on a de novo basis or via parental transmission (Reference Crusi and Engel2–Reference Bertini, Valetto, Uccelli, Tarantino and Simi4). This cytogenetic abnormality is in approximately half of the cases accompanied by developmental delay and/or learning disabilities. Its characteristic phenotype comprises, among others prominent nasal bridge, downward slanting palpebral fissures and large ears. In addition, microcephaly, growth retardation, prominent forehead, long philtrum and retrognathia are frequently found Reference Ieshima, Ogasawara, Yamamoto and Kuroki(5,Reference Ferrante, Vignetti, Antonelli, Bruni, Bertasi and Chessa6).
Ring chromosome 21 [r(21)] has been associated with a variety of congenital malformations involving nearly all organ systems Reference Rope, Hinton, Spicer, Blough-Pfau and Saal(7), structural brain anomalies Reference Muencke, Bone and Mitchell(8), haematological disorders Reference Pui, Williams, Scarnorough, Jackson, Price and Murphy(9) as well as hypogammaglobulinaemia resulting in frequent upper respiratory infections Reference Ohga, Nakao and Narazaki(10). Several cases have been reported of phenotypically normal individuals with reproductive failure (Reference Bertini, Valetto, Uccelli, Tarantino and Simi4,Reference Kleczkowska and Fryns11,Reference Huret, Leonard and Kanoui12). The main phenotypical characteristics of r(21) are summarized in Table 1. Virtually no information has been reported about the behavioural and cognitive phenotype, except that related to the presence of somatic anomalies such as hearing loss or motor dysfunction.
Table 1 Clinical features in the described patient compared to frequent features in patients with ring21 and compared to other patients with overlapping chromosome 21 deletions (anomalies with and without ring formation)

* Reported cases were aged before puberty.
† Non-specified abnormal genitalia.
With respect to the mechanism of r(21), it has been shown that ring formation arises from breaks in one of the chromosome arms followed by end to end reunion. This can lead to a stable or an unstable ring that contains duplicated regions of the proximal long arm, single-copy regions of a portion of the distal long arm and deletion of the terminal region of the long arm Reference McGinniss, Kazazian and Stetten(13). The variability in the phenotype of patients with r(21) is caused by different breakpoints, somatic loss of the ring, allelic variation, instability of the ring structure or duplication of genetic material on the ring Reference Crombez, Dipple, Schimmenti and Rao(14). Existing reports on patients with r(21) often include conventional karyotyping. However, with this technique, it is not possible to determine the exact size of possible gains and losses due to the ring formation.
In this report, a female patient with mild to moderate mental retardation and mild dysmorphisms is described, who was referred for challenging behaviours. After a r(21) was found using conventional karyotyping, whole genome single nucleotide polymorphism (SNP)-microarray analysis and targeted high-resolution array analysis of chromosome 21 were performed to fine map the aberration and to enable a reliable genotype–phenotype correlation study.
Materials and methods
Subsequent to cytogenetic analysis, whole genome array analysis was performed using a 250k SNP array according to the standard Affymetrix GeneChip protocol (Affymetrix Inc., Santa Clara, CA, USA). For further detailed structural chromosome 21 aberration detection, a high-resolution NimbleGen HG18 chromosome 21-specific 385K array was used (B3752001-00-01; Roche NimbleGen Systems, Madison, Wisconsin, USA). The 385K average probe distance was 70 bp. DNA labelling, array hybridisation, post-hybridisation washes and scanning were essentially performed according to the manufacturer's instructions (Roche NimbleGen). The acquired images were analysed using NimbleScan V2.4 extraction software (Roche NimbleGen). For each spot on the array, the log2 Cy3/Cy5 ratio (relative intensity of the Cy3 labelled patient DNA vs. the Cy5 labelled male DNA reference pool of five healthy male individuals) was calculated using the segMNT algorithm, which also applied an automatic segment detection. A 50× averaging window was generated, resulting in 3500 bp segments for this array. Breakpoints were determined using this 3.5 kb averaging window accuracy. Data were visualized in SignalMap V1.9 software (Roche NimbleGen).
Clinical observation and results
The patient is a 30-year-old female and the second child from non-consanguineous healthy parents. She has one healthy male sibling. There is no family history of mental retardation or (neuro)psychiatric disorders. She was born dysmaturally after an uncomplicated pregnancy of 40 weeks and her birth weight was 2640 g. Postnatally, she developed cyanosis and feeding problems with difficulties in swallowing. A diagnosis of urinary tract infection, most probably based on vesico-urethral reflux, was made. Her developmental trajectory showed delayed milestones with walking from the age of 20 months. At 3 years, her speech development was restricted to some monosyllabic words. Subsequently, she was evaluated by a paediatrician for behavioural problems, recurrent upper airway infections and developmental delay. A CT scan revealed, apart from local atrophy right occipital, no abnormalities. No causative explanation for the delay could be given. At the age of 7, a bifid uvula and signs of velopharyngeal insufficiency were found as well as a mixed hearing loss due to otosclerosis.
From the age of 9, she followed special education and a logopedic training programme was started because of severe retardation of her speech development. With the Wechsler Intelligence Scale for Children Revised [WISC-R Reference Van der Steene, Van Haassen and De Bruyn(15)], a verbal and performal IQ of 67 and 78 was established, respectively. Her behaviour remained problematic, in that she was hyperactive, had restricted peer interactions and was afraid of unfamiliar persons and situations. No further etiological or diagnostic hypotheses were formulated.
At age 18, she moved to a sheltered home and was employed in non-demanding activities. After 1 year, she was re-examined because of hearing loss, visual impairment and primary amenorrhoea. Bilateral otosclerosis with malformation of malleus, incus and stapedius of the right inner ear, severe myopia (−11) and delayed puberty onset based on primary oestrogen deficiency was found. Subsequently, she was treated with oestradiol/norethisteron. Karyotyping showed ring chromosome 21.
During subsequent years living in a sheltered home, her general functioning deteriorated with impaired social interactions, slight paranoid ideation, obsessive rituals, perseverations and mood instability. As a consequence, she became increasingly suggestible and easy to manipulate resulting in problematic sexual behaviours. Psychotic phenomena, especially paranoid misinterpretations, were suggested to be present and the patient was subsequently treated with risperidone in a low dose. Because of recurrent problems in social communication and reciprocity, an additional diagnosis of Pervasive Developmental Disorder Not Otherwise Specified was established. At the age of 29, she returned to her parental home and all behavioural problems disappeared within some months. She was able to restart her elementary activities at a horse riding school.
At age 30, the patient presented with a normal speech and complaints about tension, anxieties and vague ideas of reference in non-familiar situations as well as hearing and visual problems. She was verbally competent but with limited cognitive flexibility, especially with respect to problem solving and complex planning. Neuropsychiatric evaluation was done by means of a semi-structured interview using the elements of the Comprehensive Psychiatric Rating Scale Reference Asberg and Schalling(16) that covers the whole range of psychopathology. Neuropsychological assessment used a test battery comprising the Groningen Intelligence Test [GIT2 Reference Luteijn and Barels(17)], subtests of the WISC-R, the National Adult Reading Test [NART Reference Nelson(18)], the Erzigkeit's Syndrom Kurz Test Reference Lehfeld and Erzigkeit(19) and the Bermond–Vorst Alexithymia Questionnaire Reference Vorst and Bermond(20).
At psychiatric examination, she was shy and slightly tense, and there were perseverations. No formal psychiatric symptoms could be demonstrated. Assessment of intelligence revealed unequivocal results in that with the GIT2 a total IQ of 39 was found, whereas with the NART the level of intelligence was estimated to be 57, indicating a mild to moderate mental retardation with relatively impaired expressive language abilities and adequate performal capacities. With respect to her cognitive profile, only mild dysfunctions were found, especially concerning task switching and interference, relative to the level of mental retardation. As to social cognitive functioning, a mild to moderate cognitive alexithymia was found, in that the patient poorly performed on identifying and analysing emotional experiences. In contrast, no affective alexithymia could be demonstrated.
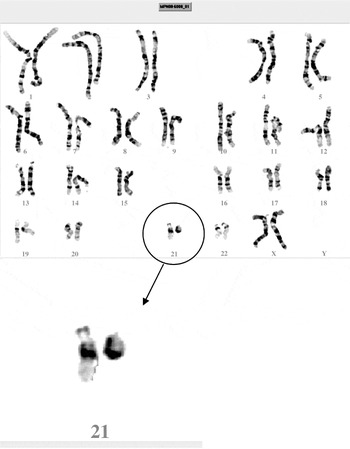
Physical examination showed a height of 161 cm and a head circumference of 54 cm. There were a severe mixed hearing loss with fixed stapedius and malformation of the malleus–incus complex right ear (AD, unchanged findings), and marked visual impairment [right eye (OD): 0.6; left eye (OS): 0.4]. The patient showed a straggle-legged walking pattern. Widespread over the skin, there were naevi pigmentosi. Detailed neurological testing demonstrated no abnormalities. Magnetic resonance imaging (MRI) scanning of the brain revealed, apart from a slight right occipital atrophia, no abnormalities. With ultrasonography of the abdomen, no malformations could be detected. Extensive laboratory tests for haematology and biochemistry demonstrated no irregularities. With respect to the phenotypical presentation, the patient showed mild facial dysmorphisms with an asymmetric face, lateral flaring of the eyebrows, low set ears, long philtrum and micro- and retrognatia (Fig. 1). In addition, she had a kyphoscoliosis, mild hypotonia, hyperlaxity of joints and pedes plani. There was clinodactyly of the right digit III and V and mild, partial cutaneous, and syndactyly of digits II and III (Fig. 2). The phenotypical characteristics of the patient, when compared with the features described in r(21) patients, are depicted in Table 1. Conventional karyotyping showed a de novo ring chromosome 21: 46,XX,der(21)r(21)(p11q22.3) (Fig. 3). Whole genome micro-array analysis confirmed the chromosome 21 aberration and did not reveal other aberrations, with the given resolution. Subsequently, targeted chromosome 21 analysis (Fig. 4) showed three separate deletions and a duplication. An interstitial deletion of 21q22.2q22.3 (40.357–45.257 Mb), an interstitial deletion of 21q22.3 (45.498–45.789 Mb) and a terminal deletion of 21q22.3 (46.447 Mb-qter). The interstitial duplication of 21q21.1 (16.196–16.280 Mb) did not comprise any genes.

Fig. 1 Facial dysmorphisms. Asymmetric face, lateral flaring of the eyebrows, low set ears, long philtrum and micro- and retrognatia.

Fig. 2 Dysmorphisms of the hands. Slender fingers, clinodactyly of digit III and V right, and mild partial cutaneous syndactyly of digits II and III.

Fig. 3 Karyotype. Ring Chromosome 21 [46,XX,der(21)r(21)(p11q22.3)].

Fig. 4 Targeted chromosome 21 analysis showed three separate deletions and a duplication: an interstitial deletion of 21q22.2q22.3 (40.357–45.257 Mb), an interstitial deletion of 21q22.3 (45.498–45.789 Mb) and a terminal deletion of 21q22.3 (46.447 Mb-qter), and the interstitial duplication of 21q21.1 (16.196–16.280 Mb) did not comprise any genes.
Discussion
In the presented female patient, a r(21) chromosome was demonstrated that was fine mapped with high-resolution targeted chromosome 21 analysis. This showed a terminal deletion, two distal interstitial deletions and a proximal gene-empty duplication on the long arm of chromosome 21. Apart from developmental delay and growth retardation, in patients with r(21) a multitude of dysmorphic features can be present. In addition, they have a predisposition for congenital malformations involving the neurologic, craniofacial, skeletal, urogenital, cardiac and haematological systems Reference Rope, Hinton, Spicer, Blough-Pfau and Saal(7). This patient had mild to moderate mental retardation, hearing loss, slightly underdeveloped sex organs with infertility and minor dysmorphisms (Table 1). In r(21) patients, the size of duplicated or deleted regions in the r(21) is assumed to be the main aspect of phenotypic variation that can range from multiple malformations to normality. However, except for a few reports (Reference Bertini, Valetto, Uccelli, Tarantino and Simi4,Reference McGinniss, Kazazian and Stetten13,Reference Falik-Borenstein, Probyl and Pulst21,Reference Ki, Rauen and Black22), r(21) patients were tested by classic cytogenetic techniques only, hampering accurate mapping of the breakpoints, resulting in a lack of reliable genotype–phenotype correlation studies. Therefore, we compared the findings in this patient to similar well-genotyped partial distal chromosome 21 deletions (chr21: 39.000 Mb-qter), including both deletions due to ring formation and due to other mechanisms (Reference Bertini, Valetto, Uccelli, Tarantino and Simi4,Reference Falik-Borenstein, Probyl and Pulst21,Reference Ehling, Kennrrknecht and Junge23,Reference Lyle, Béna and Gagos24). In concordance with these patients, she had a rather mild phenotype without severe cognitive impairment (Table 1). Behavioural problems, which were the initial referral reason in our patient, were not described in similar cases. This might be explained by the fact that, although the patient was referred for challenging behaviours and psychotic and/or autistiform symptoms, all behaviour abnormalities could be attributed to the consequences of her lower level of intellectual functioning, visual and auditory impairments, and social misinterpretations due to her disharmonic emotional profile. The latter is defined by a high level of cognitive alexithymia in the absence of affective alexithymia Reference Bermond, Vorst and Moormann(25), indicating that she was not able to identify and to verbalize her own emotional experiences, whereas her emotional sensitivity was normal.
Since the behaviour of the patient normalized completely after returning to her parent's home with a safe, well-known and structured environment, the purported presence of psychiatric symptoms has to be questioned. This corroborates the diagnostic conclusion that all challenging behaviours were contextually determined.
In conclusion, this report demonstrates the importance of the use of targeted high-resolution micro-array analysis techniques to detect small deletions and duplications in order to substantiate the genotype–phenotype correlation in patients with r(21), and stresses the significance of the recognition of cognitive alexithymia as causal factor for behavioural problems and psychiatric symptoms in patients with mental retardation in general.
Epicrisis
In the year following extensive examination, the patient gradually developed complaints about progressive fatigue that, unfortunately, were demonstrated to be caused by an acute monoblastic and monocytic leukaemia (WHO-2008: AML not otherwise specified). She was subsequently hospitalized and is following a chemotherapeutic regimen.
Acknowledgment
This study is part of a collaborative project of the research group ‘Psychopathology and Genetics' of the Radboud University Nijmegen and the Vincent van Gogh Institute for Psychiatry, Venray. All authors contributed equally to the present study. This work was supported by the AnEUploidy project, a sixth framework integrated project (B.v.B, A.H.). The authors are indebted to the patient and her parents for their kind cooperation. Figures are printed with written informed consent of the patient and her parents. The patient was referred by Mrs A. Zwaanswijk, coordinator at the Centre for Consultation and Expertise for Intellectual Disabilities, region South Holland and Zeeland.