


Short QT is a rare genetic disorder characterised by an abnormally short QT interval, with an increased risk of atrial fibrillation and sudden cardiac death. We report the case of a patient with Short QT syndrome, with mutation V141M in the KCNQ1 gene, who developed neonatal sinus bradycardia degenerating into atrial fibrillation at long-term follow-up.
Case report
A newborn female baby, born in 1994, was diagnosed with sinus node dysfunction based on the presence of sinus bradycardia and short QT interval on the electrocardiogram (Fig 1). The echocardiogram was normal. At that time, the relevance of short QT interval was underestimated, given that Short QT syndrome was first reported as a clinical entity in 2000.Reference Gussak, Brugada and Brugada 1 At 1 year of age, electrocardiogram and ambulatory electrocardiogram recordings showed sinus and junctional bradycardia at 50–60 bpm with pauses of 3.1 seconds without symptoms. A single-chamber ventricular pacemaker was implanted at 2 years of age using a transvenosus lead. No complications occurred during 7 years of follow-up, one control every 4 months. The 12-lead electrocardiogram showed a persistently short QT interval, QTc 310–270 ms during junctional rhythm at mean heart rate 50 bpm, and a clinical diagnosis of short QT was made. In 2003 and 2009, during generator replacement under general anaesthesia with sevoflurane, an electrophysiological study revealed: low-rate junctional rhythm; high-rate junctional rhythm during isoproterenol infusion; no inducible ventricular arrhythmias; and short effective refractory periods – at baseline: atrial 170/500 ms and ventricular 210/500 ms; during isoproterenol: atrial 120/350 ms and ventricular 170/350 ms.
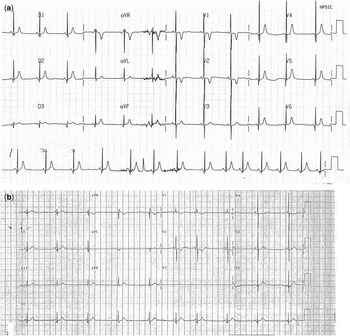
Figure 1 ( a ) Neonatal electrocardiogram showing sinus bradycardia (HR: 71 bpm, RR: 840 ms) and short QT interval (QT duration: 260 ms and QTc: 283 ms). ( b ) Infant electrocardiogram showing wandering pacemaker with intermittent junctional rhythm (HR: 54 bpm, RR: 1100 ms), QT duration 300 ms, and QTc 286 ms. T-wave amplitude is lower than that in the neonatal electrocardiogram. HR=heart rate.
At 16 years of age, during a scheduled follow-up visit, non-sustained atrial fibrillation was suspected based on the presence of irregular RR intervals with no evidence of fibrillatory waves on the electrocardiogram; 3 years later, the irregular junctional rhythm became persistent, and an electrophysiological study with three-dimensional mapping (CARTO3; Biosense Webster Inc., Diamond Bar, California, United States of America) was performed. The three-dimensional mapping showed a normal voltage map in most parts of the right atrium (Fig 2). The patient was discharged on oral anti-coagulant therapy.

Figure 2 Electroanatomical three-dimensional mapping of the right atrium during AF. Voltage map showing normal potential in most parts of the atrium, catheter positioned to record the His bundle. AF=atrial fibrillation.
Genetic analysis identified a mutation in the KCNQ1 gene (V141M), which was diagnostic for Short QT syndrome.Reference Hong, Piper and Diaz-Valdecantos 2
In the same year, the patient (50 kg) underwent electrical cardioversion after exclusion of an atrial thrombus confirmed by trans-oesophageal echocardiography, restoring sinus rhythm. Subsequently, the pacemaker system was upgraded from single to dual chamber. During in-hospital stay, hydroquinidine therapy was initiated (10 mg/kg/day in two administrations) and was continued after discharge. The QT interval did not increase significantly.
After 4 months, there was an abrupt loss of atrial sensing due to recurrent atrial fibrillation. Several unsuccessful attempts of electrical cardioversion were performed: on hydroquinidine, in wash-out, and on sotalol (4.8 mg/kg/day in three administrations).
Amiodarone therapy was initiated (200 mg/day) without any effect on QT interval and on atrial fibrillation; however, although non-sustained ventricular tachycardia was not documented by the pacemaker, in view of the high risk of life-threatening arrhythmic events between 20 and 40 years of age,Reference Mazzanti, Kanthan and Monteforte 3 amiodarone was continued. Finally, according to the recommendations based on the CHA2DS 2-VASc scoring system – 1 point due to female sex – Reference Lip, Nieuwlaat, Pisters, Lane and Crijns 4 oral anti-coagulation was discontinued and aspirin therapy was initiated.
Discussion
In 2000, Gussak et alReference Gussak, Brugada and Brugada 1 reported the first case of Short QT syndrome associated with atrial fibrillation. Gaita et alReference Gaita, Giustetto and Bianchi 5 and Giustetto et alReference Giustetto, Schimpf, Mazzanti, Maury, Scrocco and Anttonen 6 described the characteristics of Short QT syndrome, including a positive family history of sudden cardiac death, short atrial and ventricular refractory periods, and inducible ventricular fibrillation. In 2006, Giustetto et alReference Giustetto, Di Monte and Wolpert 7 reported the clinical findings and the diagnostic and therapeutic implications of Short QT syndrome. In our patient, Short QT syndrome was confirmed by electrocardiographic findings and genetic testing, consistent with those of the above-mentioned studies.Reference Gussak, Brugada and Brugada 1 , Reference Gaita, Giustetto and Bianchi 5 , Reference Giustetto, Di Monte and Wolpert 7
The first case of bradycardia and atrial fibrillation associated with Short QT and V141M mutation of the KCNQ1 gene was described by Hong et al.Reference Hong, Piper and Diaz-Valdecantos 2 Villafañe et alReference Villafañe, Fischbach and Gebauer 8 described three children with the same mutation showing low-rate atrial fibrillation at birth. Another sporadic case of this mutation was reported by Maltret et al,Reference Maltret, Wiener-Vacher and Denis 9 showing foetal bradycardia and junctional bradycardia at birth.
In our patient, during the 20 years of follow-up, different types of arrhythmias were observed. Sinus bradycardia was detected at birth, a year later the patient developed junctional bradycardia and prolonged pauses requiring prophylactic pacemaker implantation. Atrial fibrillation was observed at 16 years of age.
In contrast with previous reports,Reference Gaita, Giustetto and Bianchi 10 in our patient, hydroquinidine did not induce a significant increase in QT duration. This could be, in part, due to differences in the therapeutic doses being used.
During follow-up, no ventricular arrhythmias were observed; however, our patient is now 20 years old, an age at which the risk of sudden cardiac death is higher.Reference Mazzanti, Kanthan and Monteforte 3 The annual risk of life-threatening arrhythmias may be as high as 1.3% from the age of 20 to 40 years, whereas it is 4% in infants.Reference Mazzanti, Kanthan and Monteforte 3
We decided to continue amiodarone therapy, although there is no proof that amiodarone is effective in prolonging QT interval and preventing ventricular arrhythmias in Short QT and having the therapeutic effect on survival. According to current guidelines,Reference Priori, Wilde and Horie 11 defibrillator implantation has not been considered yet.
Conclusions
We described an asymptomatic girl with Short QT syndrome, with V141M genetic mutation in the KCNQ1 gene, who showed the evolution from neonatal sinus bradycardia, to junctional rhythm, and, finally, to low-rate atrial fibrillation, as the three faces of the same genetic problem. Owing to marked bradycardia and prolonged pauses, the patient underwent prophylactic pacemaker implantation.
Atrial fibrillation was unresponsive to multiple anti-arrhythmic agents. Electrical cardioversion was only temporarily effective.
In conclusion, we describe a V141M mutation that is associated with several arrhythmias throughout the early decades of life: sinus bradycardia, junctional rhythm, and low-rate atrial fibrillation.
Acknowledgements
The authors thank Prof. Silvia G. Priori and Dr Raffaella Bloise (Molecular Cardiology, IRCCS Fondazione Salvatore Maugeri, Pavia, Italy) for their contribution in genetic screening of the patient.
The authors also thank Dr Elisa Del Vecchio for her valuable collaboration in the editorial revision.
Financial Support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflicts of Interest
None.


