



Introduction
Spermatogenesis is a complex process that involves highly regulated cell growth and differentiation, including proliferation of spermatogonia by mitosis, genetic modifications in spermatocytes through meiosis, and differentiation of spermatids to spermatozoa with drastic morphological and functional changes to carry the genetic information to the eggs. Therefore, spermatogenesis provides an attractive model system to analyze the cellular and molecular mechanisms of developmental processes accompanying mitotic and meiotic cell divisions and cellular maturation and differentiation. Spermatogenesis is principally regulated by gonadotropins [follicle-stimulating hormone (FSH) and luteinizing hormone (LH)] and steroids (androgens, estrogens and progestins), which in turn stimulate testicular somatic cells, such as Sertoli cells and Leydig cells, to produce various molecules necessary for proper proliferation and differentiation of spermatogenic cells. Among these, cytokines are of particular interest because they are key molecules in local regulation (paracrine regulation) mediated by cell-to-cell communications (Weinbauer and Wessels, Reference Weinbauer and Wessels1999; Hedger and Meinhardt, Reference Hedger and Meinhardt2003; Oatley and Brinster, Reference Oatley and Brinster2008; Loveland et al., Reference Loveland, Klein, Pueschl, Indumathy, Bergmann, Loveland, Hedger and Schuppe2017).
Leukaemia inhibitory factor (Lif), one of the well known cytokines, exerts pleiotropic actions in various tissues. Lif maintains the undifferentiated state of mouse embryonic stem (ES) cells and allows their proliferation in vitro, although it is unnecessary for human and rabbit ES cells (Humphrey et al., Reference Humphrey, Beattie, Lopez, Bucay, King, Firpo, Rose-John and Hayek2004; Honda et al., Reference Honda, Hirose and Ogura2009). Lif also promotes the survival or proliferation of mouse primordial germ cells (PGCs) (De Felici and Dolci, Reference De Felici and Dolci1991; Chuma and Nakatsuji, Reference Chuma and Nakatsuji2001; Farini et al., Reference Farini, Scaldaferri, Iona, La Sala and De Felici2005). It is therefore plausible that a general role of Lif is to maintain the pluripotent ability and proliferation of various stem cells and their descendants, such as spermatogonial stem cells and spermatogonia in the testis. In fact, Lif has been reported to support the proliferation of rat spermatogonia under culture conditions (Dorval-Coiffec et al., Reference Dorval-Coiffec, Delcros, Hakovirta, Toppari, Jegou and Piquet-Pellorce2005), consistent with reports showing that its mRNAs are expressed in spermatogonia and somatic cells (chiefly in peritubular cells) in the rat testis (Jenab, Reference Jenab1998; Dorval-Coiffec et al., Reference Dorval-Coiffec, Delcros, Hakovirta, Toppari, Jegou and Piquet-Pellorce2005). In zebrafish, feeder cells that express Lif, fibroblast growth factor 2 (Fgf2) and glial cell line-derived neurotrophic factor (Gdnf) have been reported to enhance spermatogonial cell proliferation under culture conditions (Wong and Collodi, Reference Wong and Collodi2013). Therefore, it is likely that Lif contributes to the regulation of spermatogenesis in fish, in addition to mammals. However, the function of Lif in vivo (in the testis) is not fully understood especially in non-mammalian vertebrates, and the generality of its function in mammals and other vertebrates including fish remains to be elucidated.
Although the molecular and cellular mechanisms of spermatogenesis have been investigated by using mammals, the use of fish has some experimental advantages compared with mammals and, indeed, results obtained from studies using fish have continuously provided new insights into the regulatory mechanisms of spermatogenesis (Schulz et al., Reference Schulz, de Franca, Lareyre, Le Gac, Chiarini-Garcia, Nobrega and Miura2010). In particular, it is notable that fish spermatogenesis can be reproduced in vitro. This property facilitates analyses of the molecular mechanisms of spermatogenesis because we can directly and simply assess the effects of substances to be tested by addition to the culture medium. In vitro spermatogenesis has been reported in different fish species: eel (Miura et al., Reference Miura, Yamauchi, Takahashi and Nagahama1991, Reference Miura, Shimizu, Uehara, Ozaki, Young and Miura2011; Ohta et al., Reference Ohta, Miyake, Miura, Kamei, Aida and Miura2007), medaka (Saiki et al., Reference Saiki, Tamura, Matsumoto, Katowgi, Watanabe and Onitake1997; Shimizu et al., Reference Shimizu, Shibata and Yamashita1997, Reference Shimizu, Mita, Tamura, Onitake and Yamashita2000; Song and Gutzeit, Reference Song and Gutzeit2003; Hong et al., Reference Hong, Liu, Zhao, Xu, Wang, Liu, Chen, Deng and Gui2004; Sasaki et al., Reference Sasaki, Watanabe, Takayama-Watanabe, Suzuki, Abe and Onitake2005; Iwasaki et al., Reference Iwasaki, Ohkawa, Sadakata, Kashiwadate, Takayama-Watanabe, Onitake and Watanabe2009), zebrafish (Sakai, Reference Sakai2002; Kawasaki et al., Reference Kawasaki, Saito, Sakai, Shinya and Sakai2012; Wong and Collodi, Reference Wong and Collodi2013; Kawasaki et al., Reference Kawasaki, Siegfried and Sakai2016), tilapia (Tokalov and Gutzeit, Reference Tokalov and Gutzeit2005) and catfish (Nayak et al., Reference Nayak, Ferosekhan, Sahoo, Sundaray, Jayasankar and Barman2016). Among these, the medaka fish (Oryzias latipes) has received much attention as an experimental animal in various fields of biological science, including reproductive and developmental biology (Wittbrodt et al., Reference Wittbrodt, Shima and Schartl2002; Shima and Mitani, Reference Shima and Mitani2004), and, accordingly, various wide-ranging experimental tools have become available, including genomic and cDNA information, technical manuals, wild-type, inbred, mutant and transgenic strains, and related species belonging to the genus Oryzias [see Medaka Book, https://www.shigen.nig.ac.jp/medaka/medakabook/index.php; National BioResource Project (NBRP) Medaka, https://www.shigen.nig.ac.jp/medaka/].
Here, we report a possible role of Lif in teleost spermatogenesis. A newly established cell culture system revealed that the addition of recombinant medaka Lif proteins to the culture medium increased the number of spermatogonia. Culture of spermatogonia with Lif-overexpressing somatic cells showed similar effects. In situ hybridization and immunohistochemical analyses of the medaka testes showed that Lif proteins, as well as lif mRNAs, are expressed restrictedly in spermatogonia and Sertoli cells that surround the spermatogonia. These findings strongly suggest that Lif plays an important role in the regulation of spermatogonial cell proliferation in the medaka.
Materials and methods
Fish
Six-month-old males of hi-medaka (an orange-red variety purchased from a local fish farm) were used in this study. To visualize spermatogonia and spermatocytes, olvas–GFP medaka was also used (Tanaka et al., Reference Tanaka, Kinoshita, Kobayashi and Nagahama2001). The fish were cultured in fresh water at 27°C under artificial light conditions (14-h light and 10-h dark).
Production of baculovirus-expressed medaka Lif
Using a polymerase chain reaction (PCR)-derived Lif cDNA fragment as a screening probe, we isolated full-length Lif cDNA (DDBJ/EMBL/GenBank accession number AB766229, Supporting Information Fig. S1) from a Lambda Zap II medaka ovary cDNA library. As the cDNA contains two putative start methionines, methionine 1 (Met1) and methionine 14 (Met14), the open reading frame (ORF) from Met1 (Lif-F1) was amplified by PCR using a Topo-Lif-F1/Topo-Lif-R primer set, and the ORF from Met14 (Lif-F2) was amplified using a Topo-Lif-F2/Topo-Lif-R primer set (Fig. S1 and Table S1). The resulting ORFs were inserted into the pENTR vector by TOPO cloning (Life Technologies; Thermo Fisher Scientific, Tokyo, Japan). After confirming the sequences, the plasmids were recombined with the destination vector pET161-DEST to produce Lif proteins with a polyhistidine tag at the C-terminus (Lif-F1-His and Lif-F2-His).
ORFs encoding Lif-F1-His and Lif-F2-His were amplified from Lif cDNAs in pET161-DEST by PCR using a BamHI–Lif-F1/NotI–His primer set and a BamHI–Lif-F2/NotI–His primer set, respectively (Table S1). The resulting ORFs were cloned into the pGEM-T Easy vector (Promega, Tokyo, Japan) by TA cloning, digested with BamHI and NotI, ligated into the corresponding site of pFastBac1, and transformed into DH10Bac (Life Technologies) to obtain recombinant bacmid DNAs. Sf9 cells to which the recombinant bacmid DNA had been transfected with Cellfectin II reagent (Life Technologies) were cultured for 72 h at 28°C, and viruses producing Lif-F1-His or Lif-F2-His proteins were collected (P1 viruses). P1 viruses were further transfected into Sf9 cells for amplification and, after cultivation of the cells for 72 h, P2 viruses were collected.
Sf9 cells and their culture supernatant were collected by centrifugation 72 h after P2 virus infection. The cells were washed once in phosphate-buffered saline (PBS: 137 mM NaCl, 8.1 mM Na2HPO4 12H2O, 2.68 mM KCl, 1.47 mM KH2PO4), sonicated in extraction buffer (100 mM β-glycerophosphate, 20 mM HEPES, 15 mM MgCl2, 5 mM EDTA, 1 mM dithiothreitol, 3 µg/ml leupeptin, pH 7.5), and centrifuged to clarify the extract before immunoblotting. The presence of Lif-F1-His and Lif-F2-His proteins in the culture supernatant and cell extract was confirmed by immunoblotting as described below. As purification of Lif-F1-His or Lif-F2-His proteins from the culture supernatant or cell extract was difficult, the culture supernatant from Sf9 cells was used to examine the effect of Lif-F1-His or Lif-F2-His on spermatogonia. As a control, we also produced glutathione-S-transferase (GST)-expressing Sf9 cells by a method similar to that for medaka Lif. The culture supernatant of Lif- or GST-producing Sf9 cells was added to the spermatogonial cell culture in the ratio of 1:1 to adjust the protein concentration of Lif or GST to 250 µg/ml in the culture medium.
Characterization of Mtp1 cells
Mtp1 is a testicular somatic cell line derived from p53 (a tumour-suppressor gene)-knockout medaka (Kawasaki et al., Reference Kawasaki, Saito, Mitsui, Ikawa, Yamashita, Taniguchi, Takeda, Mitani and Sakai2009; Taniguchi et al., Reference Taniguchi, Takeda, Furutani-Seiki, Kamei, Todo, Sasado, Deguchi, Kondoh, Mudde, Yamazoe, Hidaka, Mitani, Toyoda, Sakaki, Plasterk and Cuppen2006). Its phagocytosis activity was examined as described previously (Sakai, Reference Sakai2002). We also examined expression of marker genes for Sertoli cells (gsdf, sox9b), Leydig cells (p45011β, 11βHSD2) and germ cells (olvas) in Mtp1 cells. Following isolation of total RNA samples from the testis and Mtp1 cells with ISOGEN (Nippon Gene, Tokyo, Japan), cDNAs were produced with a SuperScript III First Strand Synthesis System (Life Technologies) and analyzed by reverse transcription PCR (RT-PCR) as described previously (Takahashi et al., Reference Takahashi, Kotani, Katsu and Yamashita2014). The primer set for gsdf was 5´-CTCAGGTTGGTCAAATGAGG-3´ and 5´-CCATCATCTGAAGGTTACTTGG-3´. The primer sets for sox9b, p45011β, 11βHSD2 and olvas have been described previously (Kurokawa et al., Reference Kurokawa, Saito, Nakamura, Katoh-Fukui, Ohta, Baba, Morohashi and Tanaka2007).
Production of Mtp1 cells overexpressing medaka Lif
Exogenous genes can be transfected into mammalian and fish cells, as well as insect cells, by a baculovirus (Leisy et al., Reference Leisy, Lewis, Leong and Rohrmann2003; Yan et al., Reference Yan, Du, Chen, Yi, Li, Wang, Li and Hong2009; Yokoo et al., Reference Yokoo, Fujita, Nakajima, Yoshimizu, Kasai, Asano and Bando2013). We noticed that baculovirus-mediated gene transfer to medaka cells was less harmful than electroporation- or retrovirus-mediated methods. To produce cells that overexpress medaka Lif continuously, we therefore used a baculovirus-mediated transgenic system that employed two baculovirus vectors, one encoding the piggyBac transpose (a helper plasmid) and the other consisting of the piggyBac inverted terminal repeats (ITRs) flanking a fusion of the cytomegalovirus (CMV) promoter and the medaka Lif-F2-His cDNA (a donor plasmid) (Abe et al., Reference Abe, Miyake, Nishijima, Fujita, Sahara, Asano and Bando2005; Mizushima and Nagata, Reference Mizushima and Nagata1990; Tamura et al., Reference Tamura, Thibert, Royer, Kanda, Abraham, Kamba, Komoto, Thomas, Mauchamp, Chavancy, Shirk, Fraser, Prudhomme and Couble2000) (Figs S2 and S3). The resulting plasmids were introduced into the genome of Mtp1. Mtp1 cells were cultured in the presence of the helper and donor viruses for transfection, and 48 h later, hygromycin B was added to the culture medium at the final concentration of 500 µg/ml. Cell culture was continued for more than 2 weeks to select cells to which the transgene was stably introduced. The culture supernatant and cell extract from the drug-selected cells were analyzed by immunoprecipitation followed by immunoblotting to confirm the expression of Lif-F1-His and Lif-F2-His (Fig. S4 A). The Lif-overexpressing Mtp1 cells, as well as the wild-type Mtp1 cells, were used as feeder cells to culture medaka spermatogenic cells (see section Cell culture). The Lif-overexpressing Mtp1 cells were also used to check the specificity of anti-Lif antibody (Fig. S4 B).
Cell sorting
Medaka testes were sterilized for 2 min in 0.5% bleach in Iwamatsu’s medaka physiological saline excluding CaCl2 (IMPS (−); 111 mM NaCl, 5.4 mM KCl, 0.6 mM MgSO4, adjusted to pH 7.3 with 0.1 M NaHCO3). After washing two times in IMPS (−), three or four testes were minced with scissors, put in a tube containing 1 ml of 1.2 mg/ml dispase (Life Technologies) and 2 mg/ml collagenase (Wako, Osaka, Japan) in IMPS (−), and incubated at 28°C for 2 h with pipetting at intervals of 20 min to dissociate the tissue into single cells. The cell suspension was filtered through a 70-µm mesh and centrifuged at 420 g for 5 min at room temperature. The pellet was suspended in a culture medium (see section Cell culture) containing 2 µg/ml of propidium iodide (PI), filtrated through 40-µm mesh, and sorted by a JSAN desktop cell sorter (Bay Bioscience, Kobe, Japan). Sheath solution, which is commonly used for cell sorting, is toxic to medaka spermatogenic cells. To remove dead cells, PI-negative cells (living cells) were sorted according to the levels of forward scatter and side scatter, which roughly represent the size and internal complexity, respectively, and collected in silicon-coated tubes (1.5 × 105 cells/tube).
The obtained cell fractions were examined by a phase-contrast microscope, and the types of cells in each fraction were identified by their morphology (Fig. S5): spermatogonia were identified by a large nucleus and prominent nucleolus, primary spermatocytes were recognized by their large size and chromatin structure characteristic of meiosis, secondary spermatocytes were identified by their intermediate size, spermatid and spermatozoa were recognized by their small size and a highly differentiated morphology equipped with a flagellum, and somatic cells were recognized by their flattened and irregular shape. To identify the cell types, we also used olvas–GFP medaka, which contain GFP-labelled spermatogonia and spermatocytes (GFP expression levels in spermatogonia being larger than those in spermatocytes) (Tanaka et al., Reference Tanaka, Kinoshita, Kobayashi and Nagahama2001). Immunocytochemistry with an antibody against the meiosis marker Sycp3 (Iwai et al., Reference Iwai, Yoshii, Yokota, Sakai, Hori, Kanamori and Yamashita2006) was also performed to identify primary spermatocytes.
Cell culture
Spermatogonia-rich fractions obtained by cell sorting were co-cultured with Mtp1 or Lif-overexpressing Mtp1 cells for 7 or 8 days in a gelatin-coated plastic dish (1.5 × 105 cells per well of a 24-well plate) at 28°C. The culture medium consisted of Leibovitz L-15 (Life Technologies) supplemented with 1.7 mM proline, 0.1 mM aspartic acid, 0.1 mM glutamic acid, 0.5% bovine serum albumin, 1 µg/ml bovine insulin, 50 U/ml penicillin, 50 µg/ml streptomycin, 50 µg/ml kanamycin, 10% fetal bovine serum, 50 ng/ml retinol, 3% carp serum (obtained from carps purchased at a local fish farm), 10 IU/ml each of human chorionic gonadotropin, pregnant mare serum gonadotropin and porcine FSH, and 10 mM HEPES (pH 7.3). The medium was changed on days 1 and 4 to remove dead cells and spermatozoa, both of which were liberated from the bottom of the dish to the culture medium.
Spermatogenesis of PKH26-labelled spermatogonia in cell culture
To trace the fate of germ cells under culture conditions, cells in spermatogonia-rich fractions obtained by cell sorting were labelled with the fluorescent vital staining dye PKH26 (Sigma-Aldrich, St Louis, MO) according to the manufacturer’s instructions. PKH26-labelled cells (20 cells/well) were added to PKH26-unlabelled spermatogonia-rich fractions (1.5 × 105 cells/well) to maintain a sufficient amount of spermatogonia per well to promote the proliferation and differentiation of PKH26-labelled spermatogonia in cell culture. A mixture of PKH26-labelled and -unlabelled cells was cultured with Mtp1 or Lif-overexpressing Mtp1 cells in a gelatin-coated 24-well plate. On days 0, 3, 5 and 7, the cultured cells were harvested by centrifugation following treatment with 0.05% trypsin–EDTA (Life Technologies) and suspended in PBS in a 35-mm dish. After the types of cells had been identified by the morphological criteria (Fig. S7), PKH26-labelled spermatogonia, primary spermatocytes, secondary spermatocytes and spermatids/spermatozoa were counted under a fluorescence microscope using three wells at each time point to calculate the means and standard deviations. After counting living PKH26-labelled cells for each cell type, the cells were attached to a coverslip coated with 0.1% poly-l-lysine, fixed with 4% PFA/PBS for 15 min, and stained with anti-Sycp3 antibody (Iwai et al., Reference Iwai, Yoshii, Yokota, Sakai, Hori, Kanamori and Yamashita2006) to confirm the number of anti-Sycp3-positive cells (primary spermatocytes).
Section in situ hybridization
RNA probes were prepared by in vitro transcription with SP6 or T7 RNA polymerase using a digoxigenin (DIG) RNA-labelling Kit (Roche Diagnostics, Tokyo, Japan). Medaka testes were fixed in 4% PFA/PBS overnight at 4°C, dehydrated in ethanol, and embedded in paraffin. Sections (7 µm in thickness) were successively treated with 0.3% Triton X-100 in PBS, 0.2 N HCl, and 1 µg/ml proteinase K in PBS at 37°C and re-fixed with 4% PFA in PBS (each for 5 min). After hybridization with DIG-labelled sense or antisense probes, samples were incubated with anti-DIG-HRP antibody at 1:500 dilution (Roche Diagnostics) for 30 min. Following amplification of signals with tyramide-DNP by a TSA Plus Fluorescence Kit (PerkinElmer Japan, Yokohama, Japan), the samples were incubated with anti-DNP-Alexa Fluor 488 antibody at 1:500 dilution (Molecular Probes; Thermo Fisher Scientific, Tokyo, Japan) overnight at 4°C, stained with 10 µg/ml Hoechst 33258 for 10 min to visualize nuclei, mounted with a Prolong Anti-Fade Kit (Life Technologies), and observed under a Zeiss LSM-DUO confocal laser microscope.
Production of anti-medaka Lif antibodies
Anti-medaka Lif antibodies were raised by injecting recombinant medaka Lif proteins into guinea pigs. The recombinant proteins were produced as follows. A cDNA fragment encoding Lif-F3 was amplified by a primer set of Topo-Lif-F3 and Topo-Lif-R (Fig. S1 and Table S1). The amplified cDNA was inserted into the pENTR/D-TOPO Gateway vector with a pENTR Directional TOPO Cloning Kit (Invitrogen; Thermo Fisher Scientific, Tokyo, Japan), and the resulting plasmids were recombined with the destination vector pDEST15 using a Gateway cloning system (Life Technologies) to produce proteins with a GST tag at the N-terminus (GST-Lif-F3). GST-Lif-F3 was expressed in Escherichia coli and purified by SDS-PAGE followed by electro-elution in Tris–glycine buffer without SDS, according to the method described previously (Hirai et al., Reference Hirai, Yamashita, Yoshikuni, Lou and Nagahama1992). The antigenic proteins were injected into guinea pigs, and the antisera were affinity-purified with GST-Lif-F3 as described previously (Takahashi et al., Reference Takahashi, Kotani, Katsu and Yamashita2014). The anti-GST-Lif-F3 guinea pig antibody works in immunoprecipitation and immunocytochemistry but not in immunoblotting, probably because the antibody can recognize the native epitopes but not those modified by SDS.
Immunoprecipitation, immunoblotting and immunostaining
Immunoprecipitation and immunoblotting were performed according to the procedures described previously (Yamashita et al., Reference Yamashita, Yoshikuni, Hirai, Fukada and Nagahama1991; Ota et al., Reference Ota, Kotani and Yamashita2011) using anti-GST-Lif-F3 guinea pig antibody (this study) and anti-His G-18 rabbit antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), respectively. For immunostaining, medaka testes were fixed with 4% PFA/PBS overnight at room temperature. Following dehydration with ethanol and benzene, samples were embedded in paraffin and cut into 7-µm-thick sections. After deparaffinization and rehydration, the slides were immersed in 1 mM EDTA (pH 8.0) and heated in a microwave oven for 10 min. Following overnight incubation with anti-GST-Lif-F3 guinea pig antibody at 1:200 dilution, the slides were treated with Alexa 546-conjugated anti-guinea pig IgG antibody (Life Technologies). The expression of GFP in olvas–GFP medaka testes was immunologically detected by anti-GFP mouse antibody (Roche Diagnostics) and Alexa 488-conjugated anti-mouse IgG antibody. Immunostaining with anti-Sycp3 antibody was performed as described previously (Iwai et al., Reference Iwai, Yoshii, Yokota, Sakai, Hori, Kanamori and Yamashita2006).
Results
Characterization of baculovirus-produced medaka Lif proteins
We produced recombinant medaka Lif proteins using a baculovirus expression system. As two start methionines were presumed from its cDNA sequence (Fig. S1), two versions of Lif proteins (Lif-F1-His and Lif-F2-His) were produced. Immunoblotting analysis following immunoprecipitation showed that cell extracts from Lif-F1-His-expressing Sf9 cells contained 30-kDa and 25-kDa proteins, whereas those from Lif-F2-His-expressing cells contained 28-kDa and 25-kDa proteins. In contrast, only the 25-kDa protein was found in both of the culture supernatants (Fig. S4 A). According to the molecular masses, it is conceivable that the 30-kDa and 28-kDa proteins are full-length Lif-F1-His and Lif-F2-His, respectively. As Lif is functionally activated (matured) by removal of a signal peptide from the N-terminus (Fig. S1), the 25-kDa protein is probably a mature Lif-His. Therefore, it is most likely that a single mature Lif-His protein is produced from both Lif-F1-His and Lif-F2-His and secreted to the culture medium. The finding described below that the culture supernatants of Lif-F1- and Lif-F2-producing cells showed increases in the number of spermatogonia to similar extents supported this idea.
Spermatogonia-rich fractions used for cell culture
To examine the effects of recombinant medaka Lif proteins on spermatogenesis in cell culture, we obtained a cell fraction rich in spermatogonia. Cell sorting based on the levels of forward scatter and side scatter yielded four fractions, G-I to G-IV (Fig. 1 A). Morphological identification of cells in the fractions revealed that fractions G-I, G-II, G-III and G-IV are rich in spermatids/spermatozoa, secondary spermatocytes/spermatogonia, primary spermatocytes and spermatogonia, respectively (Fig. 1 B). We estimated that Fraction G-IV contains 10% somatic cells, 15% primary spermatocytes and 75% spermatogonia. To assess the cell population in Fraction G-IV more accurately, we specifically detected primary spermatocytes by anti-Sycp3 (a meiosis-specific protein) immunocytochemistry and found that they accounted for about 15%. An experiment using the olvas–GFP medaka that has GFP-labelled spermatogonia and spermatocytes indicated that the percentage of spermatogonia and spermatocytes is about 90% in Fraction G-IV (Fig. 1 C). These results confirmed that the ratio of spermatogonia in Fraction G-IV is about 75%. We used this fraction to evaluate the effect of Lif on spermatogenesis.
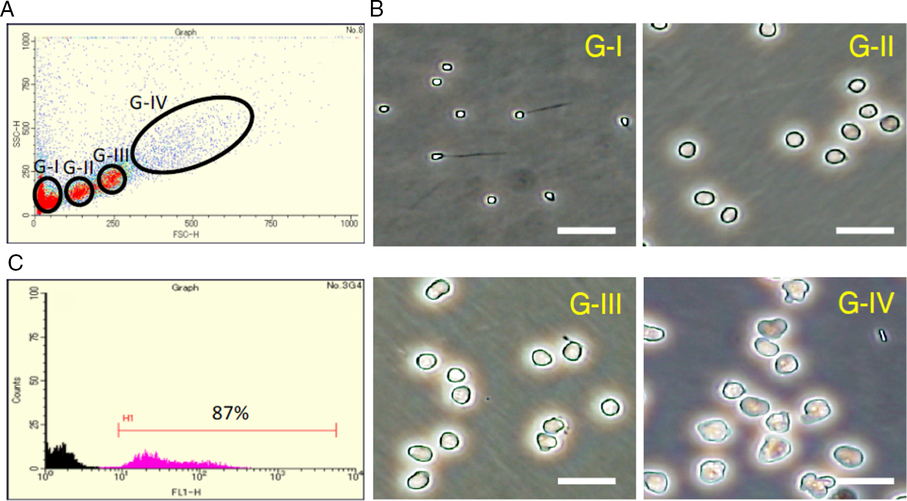
Figure 1. Cell sorting of medaka testicular cells. (A) Sorting of medaka testicular cells according to the levels of forward scatter (FSC-H) and side scatter (SSC-H). Four fractions (fractions G-I to G-IV) were obtained. (B) Phase-contrast microscopic observation of cells present in each fraction. Scale bar, 20 µm. According to the cell morphology, fraction G-I is rich in spermatozoa and spermatids (The haploid DNA content of cells in fraction G-I was confirmed by DNA fluorescence.), fraction G-II is rich in secondary spermatocytes and spermatogonia (probably at the late stage), fraction G-III is rich in primary spermatocytes, and Fraction G-IV is rich in spermatogonia (probably at the early stage). (C) The number (counts) and level of GFP fluorescence (FL1-H) of cells in Fraction G-IV obtained from wild-type (black) and olvas–GFP (magenta) medaka. Spermatogonia and spermatocytes are labelled with GFP in the olvas–GFP medaka. In Fraction G-IV, about 90% of the cells expressed GFP when the olvas–GFP medaka was used for cell sorting (H1), indicating that spermatogonia and spermatocytes account for 90% of cells in this fraction.
Cell culture system using testicular somatic cells
Our previous cell culture system without the use of feeder cells recapitulates only the later process of medaka spermatogenesis from primary spermatocytes to spermatozoa (Shimizu et al., Reference Shimizu, Shibata and Yamashita1997, Reference Shimizu, Mita, Tamura, Onitake and Yamashita2000). As testicular somatic cells, such as Sertoli cells and Leydig cells, are crucial for proper proliferation and differentiation of spermatogenic cells, we established a new cell culture system using a cell line, Mtp1 (Kawasaki et al., Reference Kawasaki, Saito, Mitsui, Ikawa, Yamashita, Taniguchi, Takeda, Mitani and Sakai2009), derived from medaka testicular somatic cells as feeder cells in expectation that this system would recapitulate the whole process of medaka spermatogenesis from spermatogonia to spermatozoa.
We characterized Mtp1 cells by their phagocytosis activity and gene expression patterns. When Mtp1 cells were cultured in the presence of polystyrene beads, about 30% of them engulfed the beads, indicating that some of the Mtp1 cells have phagocytosis activity (Fig. 6S A), a characteristic of Sertoli cells. In addition, RT-PCR analysis showed that Mtp1 cells express Sertoli cell (gsdf and sox9b) and Leydig cell (p45011β and 11β HSD2) marker genes but not a germ cell marker gene (olvas) (Fig. 6S B). These findings indicate that Mtp1 cells are heterogeneous, consisting at least of Sertoli cells and Leydig cells. The newly established cell culture system using Mtp1as feeder cells recapitulates the whole process of medaka spermatogenesis: spermatogonia proliferate and differentiate into spermatocytes, which in turn differentiate into spermatids and spermatozoa in vitro (Fig. S7). The resulting spermatozoa allow the eggs to undergo normal embryonic development after insemination.
Increase in the number of spermatogonia in the presence of recombinant Lif
Spermatogonia-enriched fractions obtained by cell sorting were cultured in the presence of Mtp1 feeder cells, and the culture supernatant of Lif-overexpressing Sf9 cells or that of GST-overexpressing Sf9 cells as a control was added to the medium. Results clearly showed that irrespective of the use of Lif-F1 or Lif-F2, the number of spermatogonia significantly increased in the presence of recombinant Lif proteins (Fig. 2 A, B).
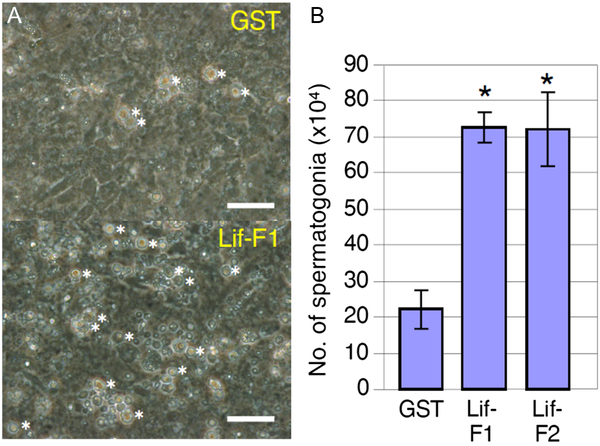
Figure 2. Effects of Lif on spermatogonia under culture conditions. (A) Culture of spermatogonia-rich fractions in the presence of baculovirus-produced GST as a control or baculovirus-produced medaka Lif proteins, Lif-F1 and Lif-F2, for 4 days. Spermatogonia identified by their morphology (see Fig. S5) were counted. The number of spermatogonia (indicated by asterisks) was increased at a higher rate in the presence of Lif-F1 than in the presence of GST. Lif-F2 had a similar effect (data not shown). Scale bar, 10 µm. (B) Number of spermatogonia in the culture on day 7 in the presence of GST or Lif (added to the culture on day 3). The number of cells at the beginning of culture (on day 0) was 15 × 104. Recombinant Lif proteins (Lif-F1, Lif-F2) increased the number of spermatogonia significantly compared with the control (GST) (mean ± standard deviation (SD); n = 3; *, P < 0.01; Student’s t-test).
Dynamics of spermatogenic cells in the presence of Lif
The increase in spermatogonia in the presence of Lif strongly suggests that Lif plays an important role in the control of spermatogonial cell proliferation. To examine the dynamics of spermatogenic cells in vitro, we performed PKH26 label and chase experiments, in which PKH26-labelled spermatogonia were co-cultured with Mtp1 or Lif-overexpressing Mtp1 cells. The number of PKH26-labelled cells (spermatogonia, primary spermatocytes, secondary spermatocytes and spermatids/spermatozoa) was examined on days 0, 3, 5 and 7 for each cell type (Fig. 3).
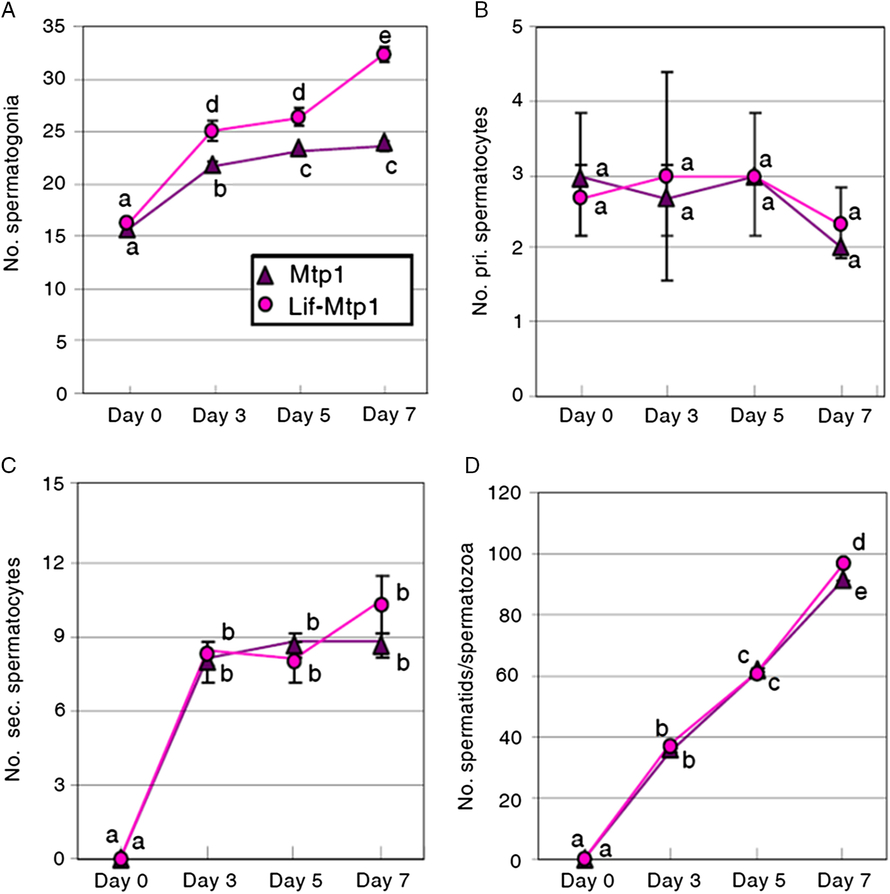
Figure 3. Dynamics of PKH26-labelled cells co-cultured with Mtp1 or Lif-overexpressing Mtp1 (Lif-Mtp1) cells. Cells were harvested on days 0, 3, 5 and 7 and PKH26-labelled cells were counted after identification of cell types by their morphology (A, spermatogonia; B, primary spermatocytes; C, secondary spermatocytes; D, spermatids/spermatozoa; see also Figs S5 and S7). The data were obtained by three independent experiments. a–eValues with different letters are significantly different (mean ± SD; n = 3; P < 0.01, Student’s t-test).
The number of PKH26-labelled spermatogonia cultured with Lif-overexpressing Mtp1 cells increased during the culture, with a higher rate of increase than those cultured with wild-type Mtp1 cells (Fig. 3 A). PKH26-labelled primary spermatocytes, the cell type of which was confirmed by anti-Sycp3 antibody, were observed on day 0 (Fig. 3 B), because of their contamination in the spermatogonia-rich fraction. In striking contrast with spermatogonia, the number of PKH26-labelled primary spermatocytes did not change significantly during the culture period from days 0 to 7, with no significant differences between the cultures with Mtp1 and Lif-overexpressing Mtp1 cells (Fig. 3 B). PKH26-labelled secondary spermatocytes were absent on day 0. They increased during the early period of culture (until day 3), but the increasing rate later reached a plateau (Fig. 3 C), probably because of an appropriate balance between the increase caused by differentiation of primary spermatocytes into secondary spermatocytes and the decrease by the differentiation of secondary spermatocytes into spermatids/spermatozoa. No significant differences in the number of PKH26-labelled secondary spermatocytes were observed between Mtp1 and Lif-overexpressing Mtp1 cells (Fig. 3 C). PKH26-labelled spermatids/spermatozoa, the end product of spermatogenesis, were not found on day 0. They continued to increase during the cultures, with no significant differences between Mtp1 and Lif-overexpressing Mtp1 cells except for day 7 (Fig. 3 D).
mRNA and protein expression of Lif in the medaka testis
As in vitro experiments demonstrated that Lif contributes to the regulation of spermatogonial cell proliferation, we decided to study the function of Lif in vivo. To this end, we first examined the expression of lif mRNA in the medaka testis by section in situ hybridization. Positive signals were found in the peripheral regions of the testis, where spermatogonia and surrounding Sertoli cells exist (Fig. 4 A, B). Closer observations confirmed that spermatogonia and Sertoli cells surrounding them express lif mRNAs (Fig. 4 C).

Figure 4. lif mRNA expression in medaka testes. Histological sections of wild-type medaka testes were subjected to in situ hybridization analysis using DIG-labelled sense (A) and antisense (B) probes. The localization of probes was visualized with the TSA Plus DNP system using anti-DIG HRP antibody and anti-DNP-Alexa Fluor 488 antibody (Lif, green). The cell nuclei were stained with the DNA dye Hoechst 33258 (DNA, blue) to identify the cell type. Positive signals were detected in the peripheral region. (C) Enlargement of the positive area indicated in (B). A DNA image (DNA) and a DNA and lif mRNA merged image (DNA/Lif) are shown. The area occupied by spermatogonia and associated Sertoli cells is marked by dots, and the nuclei of spermatogonia and Sertoli cells are indicated by yellow arrows and magenta arrows, respectively. The expression of lif mRNAs was detected in spermatogonia and Sertoli cells surrounding them but not in primary spermatocytes existing outside the marked area. Scale bar, 10 µm.
We then examined Lif protein expression by immunohistochemistry with a newly produced antibody raised against medaka Lif, the specificity of which was confirmed by using Lif-overexpressing Mtp1 cells (Fig. S4B). Specific signals were found in spermatogonia and surrounding Sertoli cells (Fig. 5 A, B). To identify the Lif-expressing cells more accurately, we examined the testes of olvas–GFP medaka, in which the intensity of GFP signals of spermatogenic cells is higher in the following order: type A (undifferentiated) spermatogonia, type B (differentiated) spermatogonia, primary spermatocytes, and secondary spermatocytes (no signals in spermatids, spermatozoa and somatic cells) (Tanaka et al., Reference Tanaka, Kinoshita, Kobayashi and Nagahama2001). Triple staining with anti-Lif antibody, anti-GFP antibody and Hoechst 33258 showed that the signal intensity of Lif corresponded well to that of GFP (Fig. 5 C), indicating that the protein levels of Lif in type A spermatogonia are higher than those in type B spermatogonia. These results are consistent with results obtained by in situ hybridization analyses (Fig. 4).
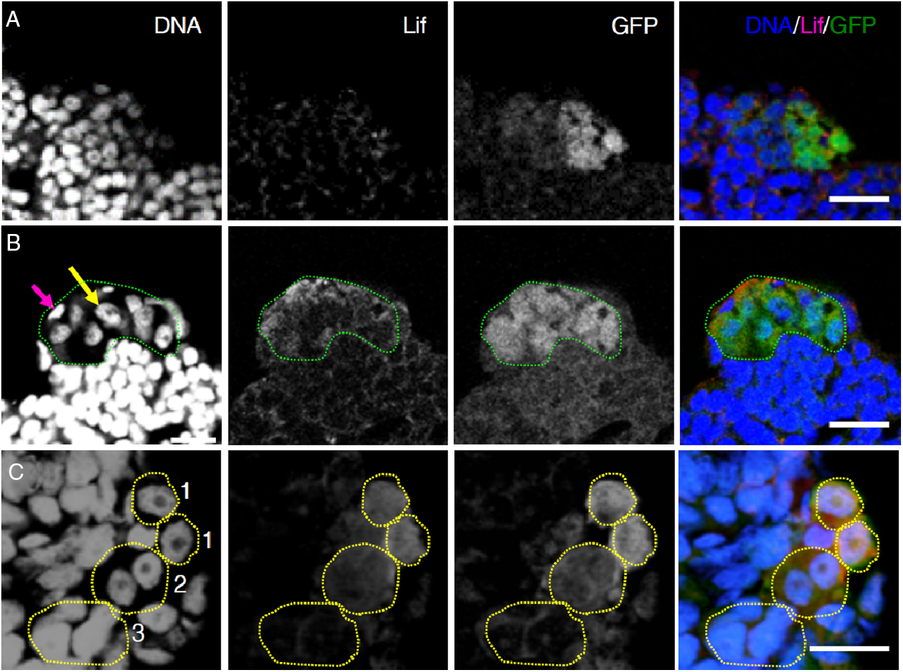
Figure 5. Lif protein expression in medaka testes. Histological sections of the olvas–GFP medaka testis triple-stained with the DNA dye Hoechst 33258 (DNA, blue), anti-Lif antibody (Lif, red) and anti-GFP antibody (GFP, green). Scale bar, 10 µm. (A) A control experiment using an antigen-absorbed anti-Lif antibody. Punctuated signals scattered all over the testis are non-specific signals due to Alexa-conjugated anti-guinea pig IgG secondary antibody. (B) Triple staining (DNA/Lif/GFP) of the olvas–GFP medaka testis. The area occupied by Lif-expressing spermatogonia (type A spermatogonia judged by their high GFP expression levels) is encircled by green dots. A yellow arrow shows the nucleus of a type A spermatogonium and a magenta arrow shows the nucleus of a Sertoli cell. The expression of Lif proteins was detected in spermatogonia and surrounding Sertoli cells. (C) Enlargement of the olvas–GFP medaka testis. Spermatogonia are characterized by a prominent nucleolus that is not stained with Hoechst 33258. GFP expression levels are higher in the following order: type A spermatogonia, type B spermatogonia and primary spermatocytes. According to their nuclear morphology and GFP expression levels, the cells enclosed by dots can be identified as type A spermatogonia (1), type B spermatogonia (2) and primary spermatocytes (3). Lif protein expression levels in type A spermatogonia are higher than those in type B spermatogonia and no expression is seen in primary spermatocytes.
Discussion
We investigated the function of Lif in medaka spermatogenesis. We found that addition of baculovirus-produced Lif to the culture medium or co-culture with Lif-overexpressing testicular somatic cells, Mtp1 cells, increases the number of spermatogonia (Figs 2 and 3). Lif proteins (Fig. 5), as well as its mRNAs (Fig. 4), are expressed in spermatogonia and surrounding Sertoli cells, with higher expression levels in type A (undifferentiated) spermatogonia than in type B (differentiated) spermatogonia. These results strongly suggest that Lif plays a critical role for the paracrine and/or autocrine regulation of spermatogonial cell proliferation in the medaka.
Taking advantage of a cell culture system that recapitulates the process of medaka spermatogenesis from spermatogonia to functional spermatozoa in vitro, we examined the behaviour of spermatogenic cells co-cultured with Mtp1 or Lif-overexpressing Mtp1 cells. Results of PKH26 label and chase experiments revealed that the increase in the number of spermatogonia is enhanced by Lif (Fig. 3 A). This finding strongly suggests that Lif regulates the proliferation of spermatogonia. How does Lif regulate spermatogonia? The number of spermatogonia can be increased by the following events if we do not take cell death or cell survival into account: (1) increase in the production rate and/or self-multiplication rate of spermatogonia; and (2) inhibition of the differentiation of type B spermatogonia into primary spermatocytes (inhibition of entry into meiosis). The numbers of PKH26-labelled primary spermatocytes were not significantly different in Mtp1 and Lif-overexpressing Mtp1 cells (Fig. 3 B), and the number of their descendants (secondary spermatocytes and spermatids/spermatozoa) increased irrespective of Lif (Fig. 3 C, D). These findings indicate that spermatogenesis continuously proceeds even in the presence Lif, thereby contradicting the possibility that the differentiation of type B spermatogonia into primary spermatocytes is inhibited by Lif.
The number of PKH26-labelled spermatogonia co-cultured with Lif-overexpressing Mtp1 cells increased at a higher rate than those co-cultured with wild-type Mtp1 cells (Fig. 3 A). In contrast, the number of PKH26-labelled primary spermatocytes neither increased significantly during the cultures nor showed differences between the two conditions (Fig. 3 B). In addition, the number of PKH26-labelled spermatids/spermatozoa, the end product of spermatogenesis, did not show apparent differences between Mtp1 and Lif-overexpressing Mtp1 cells (Fig. 3 D). These findings suggest that although Lif promotes the proliferation of spermatogonia, the resulting ‘extra’ spermatogonia do not enter meiosis on schedule (after undergoing 9 or 10 mitotic divisions in vivo) (Shibata and Hamaguchi, Reference Shibata and Hamaguchi1988). It is uncertain at present whether the ‘extra’ spermatogonia enter meiosis behind schedule (after mitotic divisions more than 10 times) or whether they disappear due to apoptosis. Culture of PKH26-labelled cells for a longer time might provide an answer to this question, although it is technically difficult at present and we need to improve the culture conditions. In the case of delayed entry into meiosis, the number of PKH26-labelled spermatids/spermatozoa will become larger in the presence of Lif according to a longer culture period. Regarding this question, it is important to examine whether Lif is involved in the survival of spermatogonia, because our present data do not exclude the possibility that the main function of Lif is to improve germ cell survival rather than to promote their proliferation. To verify this possibility, we need to analyze the apoptosis of germ cells cultured for a longer time in the presence and absence of Lif.
On the basis of the results obtained from tissue or cell culture experiments and gene knockout experiments in mammals (Stewart et al., Reference Stewart, Kaspar, Brunet, Bhatt, Gadi, Köntgen and Abbondanzo1992; De Miguel et al., Reference De Miguel, De Boer-Brouwer, Paniagua, van den Hurk, De Rooij and Van Dissel-Emiliani1996; Jenab, Reference Jenab1998; Piquet-Pellorce et al., Reference Piquet-Pellorce, Dorval-Coiffec, Pham and Jegou2000; Dorval-Coiffec et al., Reference Dorval-Coiffec, Delcros, Hakovirta, Toppari, Jegou and Piquet-Pellorce2005; Kanatsu-Shinohara et al., Reference Kanatsu-Shinohara, Inoue, Ogonuki, Miki, Yoshida, Toyokuni, Lee, Ogura and Shinohara2007; Mirzapour et al., Reference Mirzapour, Movahedin, Tengku Ibrahim, Koruji, Haron, Nowroozi and Rafieian2012; Curley et al., Reference Curley, Milne, Smith, Atanassova, Rebourcet, Darbey, Hadoke, Wells and Smith2018), Lif is thought to be required for Sertoli cells to maintain the normal spermatogenesis through regulation of spermatogonial proliferation and/or survival in mammals. As it has been reported that zebrafish feeder cells expressing Lif, Fgf2 and Gdnf enhance spermatogonial cell proliferation in cultures (Wong and Collodi, Reference Wong and Collodi2013), Lif is likely to function not only in mammalian spermatogenesis but also in teleost spermatogenesis. However, the expression and localization of Lif in the testes have not been fully examined in any vertebrate. To our knowledge, this is the first report showing the expression patterns of Lif in vertebrate testes that were determined by in situ hybridization analysis and immunohistochemistry. The finding that Lif is expressed in spermatogonia and Sertoli cells surrounding them in the medaka testes (Figs 4 and 5) strongly suggests that Lif-mediated paracrine and autocrine signals function among these cells in vivo. Further studies including studies to characterize Lif receptors and downstream signal transduction pathways consisting of JAKs and STATs (Haan et al., Reference Haan, Kreis, Margue and Behrmann2006; Loveland et al., Reference Loveland, Klein, Pueschl, Indumathy, Bergmann, Loveland, Hedger and Schuppe2017) will provide deeper insights into the mechanisms underlying Lif-mediated spermatogonial cell proliferation in the medaka testes. In addition, we need to elucidate the functional relationships between Lif and its upstream players, such as pituitary hormones (LH and FSH), steroid hormones (androgens, estrogens and progestins), and growth factors (activin, anti-Müllerian hormone (AMH)) and gonadal soma-derived factor (GSDF) (Schulz et al., Reference Schulz, de Franca, Lareyre, Le Gac, Chiarini-Garcia, Nobrega and Miura2010; Tang et al., Reference Tang, Chen, Wang, Yin, Li, Guo, Liu, Lin, Cheng and Liu2018), for a comprehensive understanding of the mechanism for regulating the switch from proliferation to differentiation of spermatogonia in the medaka testes.
In conclusion, a newly established cell culture system has demonstrated that the addition of baculovirus-produced recombinant medaka Lif proteins to the culture medium or co-culture with Lif-overexpressing medaka testicular somatic cells promotes spermatogonial proliferation. In situ hybridization and immunocytochemical analyses of the medaka testes have shown that mRNAs and proteins of Lif are expressed restrictedly in spermatogonia and Sertoli cells that surround the spermatogonia, with higher levels in undifferentiated spermatogonia than in differentiated spermatogonia. On the basis of these findings, we propose that Lif plays an important role in the regulation of spermatogonial proliferation in the medaka.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0967199419000558
Acknowledgements
We thank Dr Minoru Tanaka (Nagoya University, Japan) for providing olvas–GFP medaka. We also thank Drs Chiharu Sakai, Toshiharu Iwai, Ayako Takekoshi, Hideki Inagaki, Takeshi Usami, Shunichi Takeda, Yoshihito Taniguchi and Yoshitaka Nagahama for their help in the early phase of this study.
Financial support
This work was supported in part by the Programme for Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN) from the National Agriculture and Food Research Organization, Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (24657043), and National Institute of Genetics (NIG) Collaborative Research Programme to MY and by Grant-in-Aid for JSPS Fellows (25–2845) to RS.
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical standards
All animal experiments in this study were approved by the Committee on Animal Experimentation, Hokkaido University (permission nos. 08-0013 and 13-0099). All experiments were performed in accordance with relevant guidelines and regulations.







