


I. INTRODUCTION
In the system AgO-Ag-GeO2, there are nine different silver germanates reported so far; they are, with decreasing Ag/Ge ratio, Ag5GeO4 (Jansen and Linke, Reference Jansen and Linke1992a, Reference Jansen and Linke1992b), Ag4GeO4 (Linke et al., Reference Linke, Hundt and Jansen1995), Ag6Ge2O7 (Jansen, Reference Jansen1982; Linke and Jansen, Reference Linke and Jansen1996a), Ag8Ge3O10 (Linke and Jansen, Reference Linke and Jansen1996b), Ag10Ge4O13 (Jansen, Reference Jansen1982), Ag2GeO3 (Jansen, Reference Jansen1982), Ag2Ge2O5 (Jansen, Reference Jansen1982; Jansen and Standke, Reference Jansen and Standke1984), Ag2Ge4O9 (Wittmann and Modern, Reference Wittmann and Modern1965), and Ag4Ge9O20 (Wittmann and Modern, Reference Wittmann and Modern1965). Crystal structures of these silver germanates, except for Ag2GeO3, have been successfully determined. Parts of these silver germanates were confirmed to be isostructural with the corresponding silver silicates such as Ag5SiO4 (Linke and Jansen, Reference Linke and Jansen1994), Ag4SiO4 (Klein and Jansen, Reference Klein and Jansen2008), Ag6Si2O7 (Jansen, Reference Jansen1977; Linke and Jansen, Reference Linke and Jansen1996a), Ag10Si4O13 (Jansen and Keller, Reference Jansen and Keller1979),Ag2SiO3 (Jansen et al., Reference Jansen, Heidebrecht, Matthes and Eysel1991), and Ag2Si2O5 (Liebau, Reference Liebau1961). The most remarkable common feature of these structures is the relatively short silver-silver distances. Jansen proposed that such silver-silver contacts, ranging from 0.289 (atom distance in silver metal) to 0.340 nm [van der Waals contact distance by Bondi (Reference Bondi1964)], are induced by the attractive interactions between the d 10 configurated silver cations (Jansen and Linke, Reference Jansen and Linke1992b; Jansen, Reference Jansen1980, Reference Jansen1987). The silver atoms tend to aggregate to form two-dimensional assemblies in the form of ribbon or layered structure owing to the Ag(I)-Ag(I) interaction.
Recently, Ag2GeO3 crystal has been reported to demonstrate excellent performance of photocatalyst under indoor-illumination irradiation (Ouyang et al., Reference Ouyang, Kikugawa, Zou and Ye2009). In order to clarify the highly efficient photocatalysis mechanism and further improve its performance, the structural information is necessary for this material. In the present study, we prepared a Ag2GeO3 powder sample to determine the crystal structure from X-ray powder diffraction (XRPD) data using the Rietveld method.
II. EXPERIMENTAL
A. Synthesis
A sample of Ag6GeO3 was synthesized via the cation exchange method (Pugh, Reference Pugh1926; Ouyang et al., Reference Ouyang, Kikugawa, Zou and Ye2009). We first prepared the precursor Na2GeO3 from stoichiometric amounts of reagent-grade chemicals Na2CO3 (99.5%) and GeO2 (99.99%). They were well mixed and heated at 1173 K for 12 h, followed by quenching in air. The obtained Na2GeO3 sample was subsequently mixed with AgNO3 chemical (99.8%) in molar ratios of [Na2GeO3:AgNO3]=[1:4] and heated at 483 K for 40 h. The product was washed by distilled water to remove excess AgNO3 and by-product NaNO3. Finally, the resulting sample was dried at 343 K for 8 h to obtain the powder specimen, which was mainly consisting of Ag2GeO3 together with small amounts of Ag metal and Na2GeO3 .
B. Data collection
A diffractometer (X’Pert PRO Alpha-1, PANalytical B.V., Almelo, The Netherlands), equipped with an incident-beam Ge(111) Johansson monochromator to obtain Cu Kα 1 radiation and a high-speed detector, was used in the Bragg-Brentano geometry. The X-ray generator was operated at 45 kV and 40 mA. A variable divergence slit was used to keep a

Figure 1. (Color online) (Color online) Comparison of the observed diffraction pattern of Ag2GeO3, Ag, and Na2GeO3 (symbol: +) with the corresponding calculated pattern (upper solid line). The difference curve is shown in the lower part of the diagram. Vertical bars indicate the positions of possible Bragg reflections for Ag2GeO3, Ag, and Na2GeO3.
constant illuminated length of 5 mm on the specimen surface. Other experimental conditions were the following: continuous scan, experimental 2θ range from 6.0039° to 148.9353° (an accuracy of ±0.0001° 2θ), 17 107 total data points, and 17.5 h total experimental time. The structure data were standardized according to the rules formulated by Gelato and Parthé (Parthé and Gelato, Reference Parthé and Gelato1984) using the computer program STRUCTURE TIDY (Gelato and Parthé, Reference Gelato and Parthé1987). The crystal-structure models were visualized with the computer program VESTA (Momma and Izumi, Reference Momma and Izumi2008).
III. RESULTS AND DISCUSSION
A. Crystal structure refinement
The XRPD pattern in Figure 1 showed the presence of weak diffraction intensities peculiar to Ag and Na2GeO3; hence we carefully determined the peak positions with Ag2GeO3 by finding minima in the second derivatives using the computer program PowderX (Dong, Reference Dong1999). The 2θ values of 26 observed peak positions were then used as input data to the automatic indexing computer program TREOR90 (Werner et al., Reference Werner, Eriksson and Westdahl1985). One orthorhombic unit cell was found with satisfactory figures of merit: M26/F26=37/37 (0.006 174, 115) (de Wolff, Reference de Wolff1968; Smith and Snyder, Reference Smith and Snyder1979). The derived unit-cell parameters of a=0.463 10(6) nm, b=0.713 90(6) nm, and c=1.041 01(8) nmcould index all reflections with Ag2GeO3 in the observed diffraction pattern. The observed diffraction peaks were examined to confirm the presence or absence of reflections. There was no systematic absence for hkl, 0kl, h0l, nor hk0 reflections, which implied that one of the possible space groups was P212121. This space group and the derived unit-cell dimensions were in accord with those of Ag2SiO3 [space group P212121 and unit-cell dimensions a=0.4527(1) nm, b=0.7108(1) nm, and c=0.9959(1) nm], which strongly suggests that Ag2GeO3 and Ag2SiO3 are isotypic.
The initial structural parameters of Ag2GeO3 were taken from those of Ag2SiO3 (Jansen et al., Reference Jansen, Heidebrecht, Matthes and Eysel1991). There are six independent sites (i.e., two Ag site, one Ge site, and three O sites) with the Wyckoff position 4a in the unit cell. The structural parameters of all atoms were refined by the Rietveld method (Rietveld, Reference Rietveld1967) using the computer program RIETAN-FP (Izumi and Momma, Reference Izumi and Momma2007) with the profile-intensity data in the 2θ range of 13.0059° to 148.9353° (Figure 1). The structural model of Ag metal (Becherer and Ifland, Reference Becherer and Ifland1954) and that of Na2GeO3 (Cruickshank et al., Reference Cruickshank, Kalman and Stephens1978) were included in the refinement as coexisting phases. A Legendre polynomial with 12 adjustable parameters was fitted to background intensities. The split Pearson VII function (Toraya, Reference Toraya1990) was used to fit the peak profiles. The preferred-orientation parameter of March-Dollase function (Dollase, Reference Dollase1986), r, was refined to be r=0.972(2)with the preferred-orientation vector [001], suggesting that the crystal grains were slightly fractured along the cleavage planes parallel to (001). Isotropic displacement (B) parameters were assigned to all atoms. All of the B parameters of oxygen atoms were constrained to have the same value. Reliability indices (Young, Reference Young and Young1993) for a final result were Rwp=5.58%, S(=Rwp/Re)=1.26, and Rp=4.20% (RB=0.67% and RF=0.35% for Ag2GeO3). Crystal data are given in Table I
TABLE I. Crystal data for Ag2GeO3.

TABLE II. Structural parameters for Ag2GeO3.
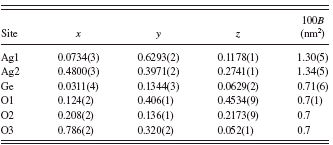
and the final atomic positional and B parameters are listed in Table II.
Quantitative X-ray analysis with correction for microabsorption according to Brindley’s procedure (Brindley, Reference Brindley1949) was implemented in the program RIETAN-FP. The phase composition was found to be 96.5 mass % Ag2GeO3, 1.2 mass % Ag, and 2.3 mass % Na2GeO3. These values are in accord with those of the potocatalyst specimen (92.6 mass % Ag2GeO3, 3.4 mass % Ag, and 4.0 mass % Na2GeO3) that was prepared in a previous study (Ouyang et al., Reference Ouyang, Kikugawa, Zou and Ye2009).
B. Structure description
Figure 2 shows the refined structural model of Ag2GeO3. The structure is isomorphous with Ag2SiO3. Selected interatomic distances and bond angles, together with their standard deviations, are listed in Table III). There are two crystallographically independent silver atoms, Ag1 and Ag2. Each Ag1 atom is coordinated by three O atoms and each Ag2 atom is almost linearly coordinated by two O atoms. These Ag atoms aggregate to form a layered structure (Figure 3, in analogy with Ag2SiO3. Individual silver layers are stacked along the [001] direction, and they are interconnected via infinite single chains of [Ge2O6] to form a three-dimensional structure.
The chains of [Ge2O6] are running parallel to [100], with two tetrahedra per identity. The mean Ge-O bond length of 0.176 nm in the GeO4tetrahedra is in good agreement with

Figure 2. (Color online) (Color online) Perspective a-axis projection of Ag2GeO3. Numbering of silver atoms corresponds to those given in Table II.
TABLE III. Bond lengths (nm) and angles (deg) for Ag2GeO3.

a Symmetry transformations used to generate equivalent atoms: −x+1/2, −y, z+1/2.
b Symmetry transformations used to generate equivalent atoms: −x, y+1/2, −z+1/2.
c Symmetry transformations used to generate equivalent atoms: x+1/2, −y+1/2, −z.
that expected from the bond valence sum (0.1748 nm). The O-Ge-O angles (the mean value=109.5°) are more distorted from the regular tetrahedral value than the O-Si-O angles in Ag2SiO3. These mean values are in good agreement with those found in other metagermanates Na2GeO3 (Cruickshank

Figure 3. (Color online) (Color online) Arrangement of silver atoms in Ag2GeO3. Numbering of atoms corresponds to those given in Table II.
et al., Reference Cruickshank, Kalman and Stephens1978), CoGeO3 (Peacor, Reference Peacor1968), and MnGeO3 (Fang et al., Reference Fang, Townes and Robinson1969). As is common for germanates and silicates, the Ge-O (bridging)-Ge angle (129.5°) in Ag2GeO3 is smaller than the Si-O-Si angle (131.4°) in Ag2SiO3. The polyhedral volume of [GeO4] (0.002 798 nm3) is larger than that of [SiO4] (0.002 230 nm3) . Because the tetrahedral chains lie between the silver layers, the interlayer spacings are larger for Ag2GeO3 than for Ag2SiO3.
It was reported that the bonding Ag(I)-Ag(I) interactions usually range from 0.289 to 0.340 nm (Jansen, Reference Jansen1980, Reference Jansen1987). The silver-silver distances within individual silver layers satisfy this requirement for both Ag2GeO3 and Ag2SiO3; they are 0.2992(2) to 0.3390(2) nm for the former (Table III) and 0.2933(1) to 0.3352(1) nm for the latter. This implies that the attractive interaction between silver atoms would be actually operative and indispensable for the stabilization of the silver substructure. On the other hand, between the silver layers, the Ag(I)-Ag(I) interaction is not expected for Ag2GeO3 but anticipated for Ag2SiO3 because the nearest silver-silver distances between adjacent layers are 0.3591(2) (>0.340) nm for the former and 0.3359(1) (<0.340) nm for the latter. Consequently, the Ag2GeO3 structure can be characterized by the absence of attractive interaction between the silver layers, although the two silver compounds are isotypic.
IV. CONCLUSIONS
We determined the crystal structure of silver metagermanate Ag2GeO3, having an orthorhombic unit cell with the space group P212121. The crystal structure consists of silver layers and interconnecting single chains of [Ge2O6]. The relatively short Ag-Ag distances within individual silver layers indicate Ag(I)-Ag(I) interaction. Although Ag2GeO3 and Ag2SiO3 are isotypic, the former structure is characterized by the absence of Ag(I)-Ag(I) interaction between the adjacent silver layers.






