I. INTRODUCTION
Although predominantly occurring as a chelating agent, ethylenediamine (for simplicity, further denoted as en) has been observed to possess the capacity to act as a bridging ligand as well. Various coordination polymers have been reported so far with en linking neighboring metal centers such as: Co(II) (Lauchan et al., Reference Lauchan, Prior, Meansiri and Rujiwatra2008), Zn(II) (Pritchard et al., Reference Pritchard, Eaton, McDonald and Strickland2001), or Ag(I) (Näther and Beck, Reference Näther and Beck2004).
Cd(II) coordination arrays with extended structures formed via small anionic bridges are numerous (Zhou et al., Reference Zhou, Xu, Yuan and Hong2003; Xia et al., Reference Xia, Hu, Dai, Wu, Fu, Zhang and Du2004; Abu-Youssef, Reference Abu-Youssef2005). Examples of en connecting Cd(II) centers, whether exclusively or together with other types of bridging ligands present, are also known in the literature (Shen et al., Reference Shen, Xu, Liu and Xu2002; Yilmaz et al., Reference Yilmaz, Yilmaz and Kazak2005).
Two polymeric compounds presented here, [Cd(en)Cl2]n and [Cd(en)Br2]n, have already been synthesized and examined by means of thermogravimetry analysis, IR and Raman spectroscopy (Iwamoto and Shriver, Reference Iwamoto and Shriver1971; De and Chaudhuri, Reference De and Chaudhuri1984). Investigations revealed the presence of bridging trans en ligands. The authors, however, having assumed cubic, tetragonal, or hexagonal lattices, failed in their attempts to index the powder diffraction patterns they obtained (and therefore never submitted them for publication). To the best of our knowledge, no one else has successfully tried to solve the crystal structures of these polymers prior to this paper.
II. EXPERIMENTAL
A. Synthesis
Poly[cadmium(II)-di-μ-chloro-μ-(ethylenediamine-N,N’)] (1) and poly[cadmium(II)-di-μ-bromo-μ-(ethylenediamine-N,N’)] (2) were obtained from aqueous solutions of 8 mmol of CdX2 (1.82 g CdCl2·H2O and 2.18 g CdBr2, respectively). After heating to 60–70 °C, 8 mmol of ethylenediamine (0.53 ml) was added dropwise to each of the solutions. White precipitates were formed immediately and were collected after a few days (aging process), washed with 2-propanol and dried at 60 °C. They were air stable and not affected by moisture.
The same synthetic procedure was applied to obtain a CdI2-based analogous polymer, but consecutive approaches yielded only bis[tris(ethylenediamine-N,N′)-cadmium] tetraiodocadmate diiodide complex, [Cd(en)3]2[CdI4]I2 (Wieczorrek and Tebbe, Reference Wieczorrek and Tebbe1998). This is in agreement with the predictions of Iwamoto and Shriver (Reference Iwamoto and Shriver1971), who argued that en bridging in CdI2 system would be energetically unfavorable.
B. Data collection
X-ray diffraction patterns of mortar-and-pestle-ground, back-loaded, pressed specimens were taken at room temperature using an X'Pert Pro MPD diffractometer (see details in Table I). Experiments were carried out in Bragg-Brentano geometry. Peak positions were determined with software from the PROSZKI package (Łasocha and Lewiński, Reference Łasocha and Lewiński1994).
Table I. Parameters of X-ray powder diffraction measurements.

Program ITO-12 (Visser, Reference Visser1969), included in the PROSZKI was used for indexing purposes. Structures were solved using the direct methods program EXPO2004 (Altomare et al., Reference Altomare, Caliandro, Camalli, Cuocci, Giacovazzo, Moliterni and Rizzi2004). Crystal structure refinement was carried out by using the JANA2006 (Petricek et al., Reference Petricek, Dusek and Palatinus2006) program. The pseudo-Voigt function was used to generate the line shape of the diffraction peaks. A total of 43 parameters were refined, including scale factor, background, zero point, unit-cell parameters, and fractional coordinates as well as those describing peak shape, width, and peak asymmetry. C–C and C–N distances in en molecules were weakly restrained to 1.54(±0.02) and 1.44(±0.04) Å, respectively. Hydrogen atoms were placed in idealized positions (by JANA2006 program assuming sp3 hybridization) and not refined.
Tabulated powder diffraction data have been sent to the International Centre for Diffraction Data (ICCD), where they have been accepted and assigned PDF numbers 00-061-11059 and 00-061-11058, respectively.
III. RESULTS AND DISCUSSION
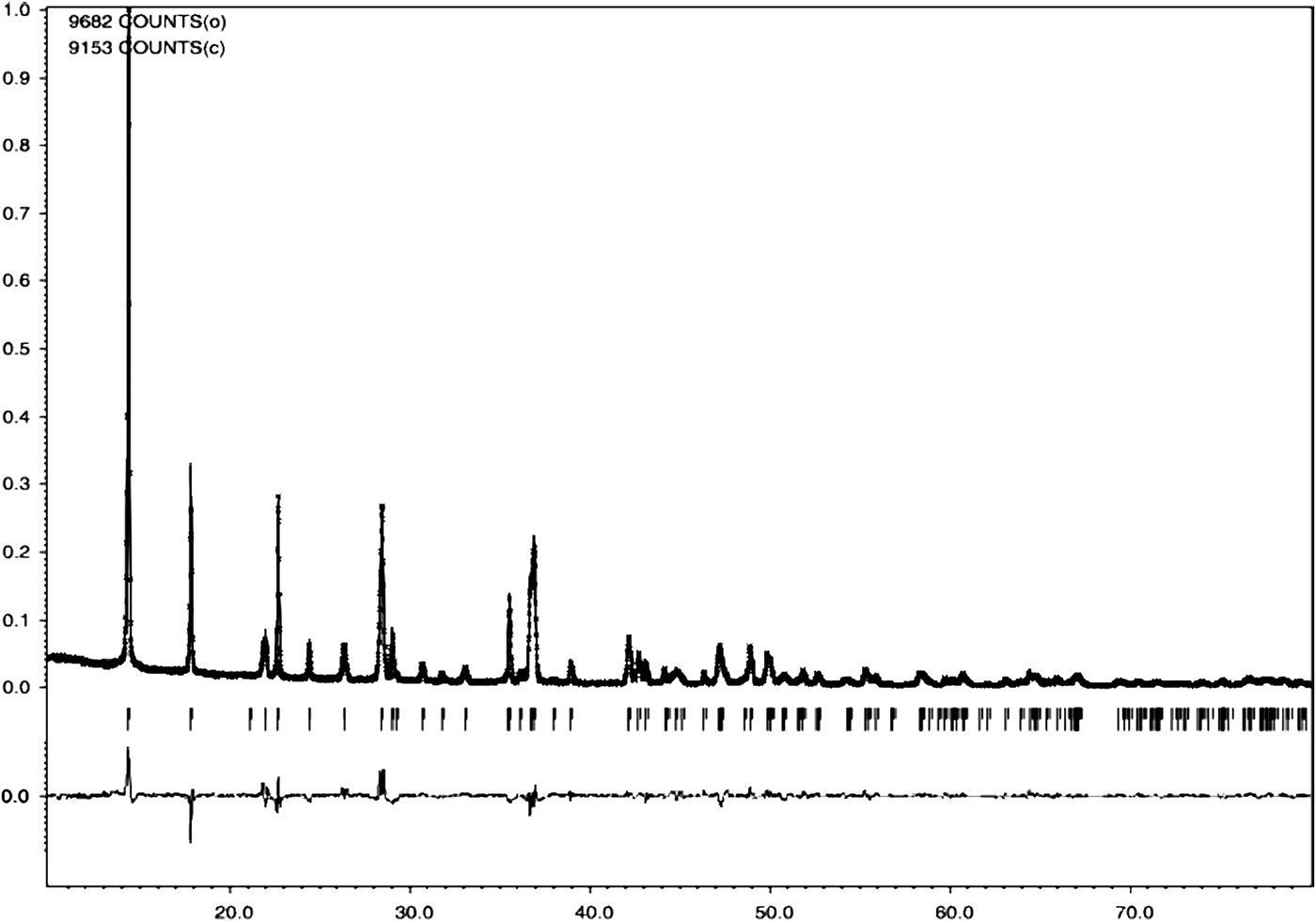
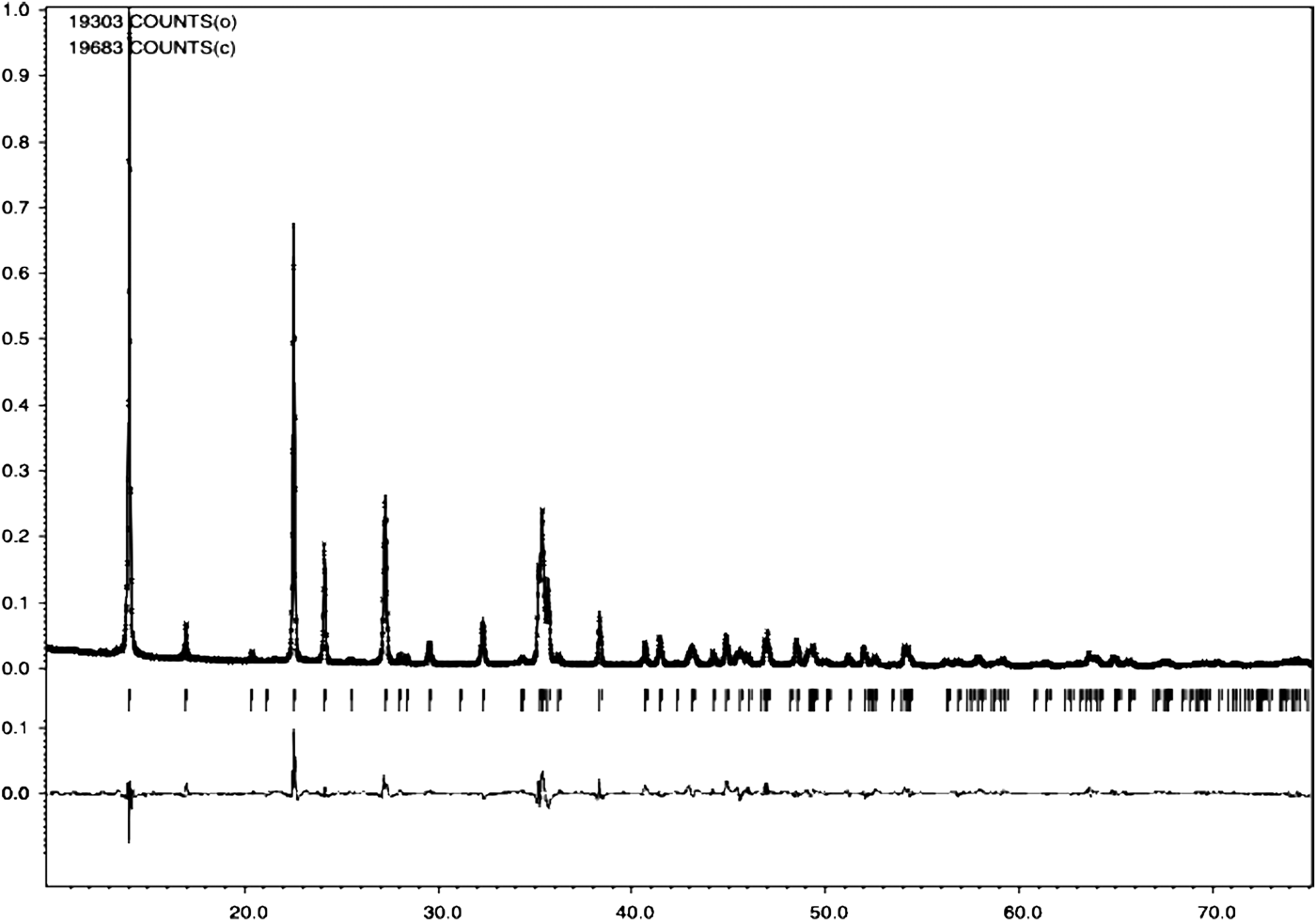
Figures 1 and 2 show the powder diffraction patterns of the investigated compounds together with the results of Rietveld analysis (peak positions, calculated, and difference curves). In both cases, Bragg reflections were indexed based on an orthorhombic unit cell; obtained unit-cell parameters and figures of merit F N (Smith and Snyder, Reference Smith and Snyder1979) are given in Table II. Both compounds were found to be isostructural, crystallizing in the space group Pbam (No. 55). There are two formula units in one unit cell of each compound. The refined structural parameters are listed in Tables III and IV and selected interatomic distances and angles in Tables V and VI.

Figure 1. Observed (black, straight line), calculated (red, crosses), and difference (blue, straight line) Rietveld profiles for (1). Green (vertical) bars indicate the positions of Bragg reflections.
Figure 2. Observed (black, straight line), calculated (red, crosses), and difference (blue, straight line) Rietveld profiles for (1). Green (vertical) bars indicate the positions of Bragg reflections.
Table II. Crystal data and structure refinement details for (1) and (2).
Table III. Atomic coordinates and isotropic displacement parameters for (1).

Table IV. Atomic coordinates, occupancies, and isotropic displacement parameters for (2).

Table V. Selected interatomic distances (Å) and angles (°) for (1).

Symmetry codes: (i) x, y, z − 1, (ii) −x + 1, −y, z − 1, (iii) −x + 1, −y, z, (iv) −x + 1, −y − 1, z
Table VI. Selected interatomic distances (Å) and angles (°) for (2).

Symmetry codes: (i) x, y, z − 1; (ii) −x + 1, −y, z − 1; (iii) −x + 1, −y, z; (iv) x, y, −z; (v) −x + 1, −y − 1, z; (vi) x, y, z + 1.
X-ray structural analysis of (1) reveals a neutral polymeric coordination compound. As shown in Figure 3, the coordination environment of each Cd(II) center consists of two N-donor atoms from en molecules in trans coordination and four chloride anions to constitute a distorted octahedron. Adjacent Cd coordination octahedra share Cl–Cl (or Br–Br) edges, thus forming one-dimensional chains spreading infinitely along the c-direction. Two en molecules interconnect Cd centers on alternate sides of these chains; as a result, two-dimensional sheets are created (see Figures 3 and 4).
Figure 3. Hybrid organic–inorganic layer of (1) showing octahedral coordination environment of Cd2+ ions. Cd = pink(octahedron centre), Cl = green (vertices), N = gray (medium), and C = black (small). The smallest circles represent hydrogen atoms.
Figure 4. Packing of 2D layers in (1). Notations of atoms are the same as in Figure 3.
It has already been stated that the chloride and bromide polymers are isostructural. Distances between neighboring Cd centers were found to be 4.042 Å in (1) and 4.185 Å in (2), more than the sum of their van der Waals radii (r vdW_Cd = 1.58 Å), thus excluding metal–metal interactions. Cd–Cl bonds of 2.703(10) Å agree well with data on Cd–halide complexes gathered by Bailey and Pennington (Reference Bailey and Pennington1997), whereas Cd–Br bonds of 2.837(7) Å are a little bit longer [Cd–Clrange: 2.576(7)–2.731(2) Å; Cd–Brrange: 2.691(1) − 2.819(2) Å]. For both polymers, Cd–N bonds of 2.43(6) Å in (1) and 2.28 (6) Å in (2) fit within the range found in similar compounds by Strasdeit et al. (Reference Strasdeit, Saak, Pohl, Driessen and Reedijk1988) [Cd–Naliphatic range: 2.25(1) − 2.44(1) Å].
The substitution of Cl by Br in (2) is responsible for lengthening of Cd–halide and slight contraction of Cd–N bonds. Similar trends were observed and reported for a series of cadmium halide complexes with other N-coordinating ligands (Strasdeit et al., Reference Strasdeit, Saak, Pohl, Driessen and Reedijk1988). These changes could indicate a tendency to lower the coordination number of cadmium from 6 to 4 because of steric hindrance, with the greater size of the anion when moving down the series, accompanied by the softer character of the heavier halide, resulting in increased electronic density at the metal centre. This chain of arguments serves, then, as another explanation of our unsuccessful attempts to obtain [Cd(en)I2]n.
IV. CONCLUSION
It is worth noting that the crystal structure solution of (1) and (2) from powder diffraction data corroborates the structural model of Iwamoto and Shriver (Reference Iwamoto and Shriver1971) of cadmium (II) halide polymers with bridging en ligands in trans coordination and illustrates in an elegant manner the complementary character of solid state investigation methods. By contemporary powder diffraction methods, interesting but quite old scientific problems concerning crystal structure of cadmium(II) diamine complexes (described so far by IR, RS, and TGA methods only), could be easily solved.
Crystal structure data have been deposited at the Cambridge Crystallographic Data Centre and allocated the deposition numbers CCDC 911610 {1} and 911611 {2}, and can be obtained on request.
ACKNOWLEDGMENT
The support of the Polish government program MNiSW, grant NN204546439, is acknowledged.