Introduction
Lichens are the symbiotic phenotype of nutritionally specialized fungi (mycobionts) that derive fixed carbon from green algae and/or cyanobacteria (Honegger Reference Honegger1991). They are distributed worldwide, inhabiting different environments. Unfortunately, our understanding of genetic variation in lichen populations is still very limited. Molecular studies could help to explore population history, mode and effectiveness of lichen dispersal and gene exchange among lichens. In a number of studies, AFLP and RAPD techniques have been employed (e.g. Murtagh et al. Reference Murtagh, Dyer, McClure and Crittenden1999, Reference Murtagh, Dyer and Crittenden2000; Dyer et al. Reference Dyer, Murtagh and Crittenden2001; Seymour et al. Reference Seymour, Crittenden, Dickinson, Paoletti, Montiel, Cho and Dyer2005; Honegger & Zippler Reference Honegger and Zippler2007; Lindblom & Ekman Reference Lindblom and Ekman2012). However, as lichens represent a bi- or tripartite symbiosis, in most cases anonymous fingerprinting is not applicable for studies of lichen populations (Walser et al. Reference Walser, Sperisen, Soliva and Scheidegger2003). Genetic markers for population studies of lichens should combine the detection of high levels of genetic variation and the selective amplification of fungal or algal DNA. The advantage of microsatellites is their high variation that can resolve genets, making such markers especially useful in studies at fine spatial scales.
Molecular approaches have also been extensively used to resolve relationships among lichen photobionts and investigate population structures of lichen-forming algae, mainly based on ITS rDNA sequencing (e.g. Beck et al. Reference Beck, Friedl and Rambold1998; Helms et al. Reference Helms, Friedl, Rambold and Mayrhofer2001; Piercey-Normore Reference Piercey-Normore2006), but recently new microsatellite markers have been reported for Dictyochloropsis reticulata, a symbiotic alga of the lichen Lobaria pulmonaria (Dal Grande et al. Reference Dal Grande, Widmer, Beck and Scheidegger2010), Trebouxia spp. from Parmotrema tinctorum (Mansournia et al. Reference Mansournia, Wu, Matsushita and Hogetsu2012) and T. decolorans from Xanthoria parietina and Anaptychia ciliaris (Dal Grande et al. Reference Dal Grande, Beck, Singh and Schmitt2013).
There are only a limited number of fungus-specific primers available, allowing detection of variation in lichen populations (e.g. Zoller et al. Reference Zoller, Scheidegger and Sperisen1999; Lindblom & Ekman Reference Lindblom and Ekman2006). Until recently, the most commonly used markers in lichen population genetics were ribosomal loci: the internal transcribed spacer (ITS), the intergenic spacer (IGS), the small subunit (SSU) and the large subunit (LSU) (for review see Werth Reference Werth2010). Microsatellites are known to be informative markers at the population level in different organisms and were previously developed and used for the lichen Lobaria pulmonaria (e.g. Walser et al. Reference Walser, Sperisen, Soliva and Scheidegger2003, Reference Walser, Gugerli, Holderegger, Kuonen and Scheidegger2004, Reference Walser, Holderegger, Gugerli, Hoebee and Scheidegger2005; Dal Grande et al. Reference Dal Grande, Widmer, Wagner and Scheidegger2012; Werth & Scheidegger Reference Werth and Scheidegger2012). Recently, microsatellite primers were also developed for the tropical Peltigera dolichorhiza complex (Magain et al. Reference Magain, Forrest, Sérusiaux and Goffinet2010), the Antarctic endemic lichen fungus Buellia frigida (Jones et al. Reference Jones, Green, Hogg and Wilkins2012) and Parmotrema tinctorum (Mansournia et al. Reference Mansournia, Wu, Matsushita and Hogetsu2012). However, the problem of specificity arises when working with symbiotic organisms. Walser et al. (Reference Walser, Sperisen, Soliva and Scheidegger2003) used manually separated fungal material, isolated from apothecia of Lobaria pulmonaria, for microsatellite development in order to reduce the risk of contamination with algal DNA. However, Widmer et al. (Reference Widmer, Dal Grande, Cornejo and Scheidegger2010) proved that five of twelve primer pairs published by Walser et al. (Reference Walser, Sperisen, Soliva and Scheidegger2003) are indeed algal-specific markers. This example shows that development of species-specific markers is very difficult for fungal associations, due to the risk of contamination of the target DNA with the symbiont's genome. The use of axenic cultures does undoubtedly facilitate biont-specific marker development.
Protoparmeliopsis muralis (Schreb.) Choisy (syn. Lecanora muralis) is a green-algal lichen that colonizes different substrata, such as calcareous and siliceous stones, wood and the bases of roadside trees. It is cosmopolitan, and very common in the Northern Hemisphere, especially in urban areas. A recent molecular study showed that P. muralis forms a strongly supported monophyletic group with other lobate species (Pérez-Ortega et al. Reference Pérez-Ortega, Spribille, Palice, Elix and Printzen2010). Thalli of this lichen usually bear many apothecia that produce numerous spores for dispersal. Although the name Protoparmeliopsis muralis is used here, we recognise that there are problems concerning its validity (see Laundon Reference Laudon2010).
Several species of Trebouxia photobionts have been reported from this lichen so far (T. asymmetrica, T. gigantea, T. cf. impressa, T. incrustata and unidentified Trebouxia sp.) (Guzow-Krzemińska Reference Guzow-Krzemińska2006). The low level of selectivity of the mycobiont, with respect to the choice of its photobiont, was postulated to be the key factor allowing Protoparmeliopsis muralis to be one of the most successful urban lichens in the world (Guzow-Krzemińska Reference Guzow-Krzemińska2006). It is generally assumed that the ability of a species to adapt and occupy new habitats is determined by its genetic variation. However, our knowledge about the intra-specific variation of this lichen-forming fungus is very limited.
Tools for population studies on lichens that are rare in Europe, such as Lobaria pulmonaria, have already been developed. However, markers for common lichens that are widely distributed and ecologically less specific are also needed. This would allow a comparison of the level of intraspecific variation and/or the mode of dispersal between rare and common lichens. The objective of this study was the development and characterization of microsatellite markers specific for the mycobiont Protoparmeliopsis muralis. Fungus-specific microsatellite markers could be particularly useful for population studies of this lichen, and to determine whether there is any genetic variation within a given lichen thallus.
Materials and Methods
Mycobiont culture
In order to avoid algal contamination, the mycobiont cultures were obtained from fungal spores using the method of Ahmadjian (Reference Ahmadjian1993), modified according to Stocker-Wörgötter (Reference Stocker-Wörgötter, Kranner, Beckett and Varma2002). At the beginning of the experiment, a pre-washing step was performed by placing the fruiting bodies in double-distilled water with a small drop of Tween 80 (detergent) on a magnetic stirrer to remove dirt particles from the surface of the apothecia. Two specimens of Protoparmeliopsis muralis were used for mycobiont isolation (BGK247 and BGK258, both collected from concrete in Salzburg, Austria). The apothecia were attached to the top cover of the Petri dish and placed over BBM (Deason & Bold Reference Deason and Bold1960; Bischoff & Bold Reference Bischoff and Bold1963) medium. The germination of the fungal spores was observed using dissecting and transmission light microscopes. The blocks of agar beneath germinating ascospores were cut out and transferred to a nutrient-rich medium containing mannitol. The mycobiont cultures were obtained from multiple spores from a single apothecium. For subculturing, small fungal colonies were homogenized with sterile double-distilled water in a mortar and the suspension was transferred with a Pasteur pipette to a new Petri dish containing nutrient medium. The mycobionts were subcultured on G-LBM (Brunauer et al. Reference Brunauer, Hager, Grube, Türk and Stocker-Wörgötter2007), Potato-Dextrose Agar (PDA) media and BBM (Deason & Bold Reference Deason and Bold1960; Bischoff & Bold Reference Bischoff and Bold1963) enriched with 0·5% mannitol. The cultures were kept in the dark in the culture chamber at 20°C for 14 h and 10°C for 10 h. Well-developed mycelia were used for further experiments.
Identification of mycelial cultures by ITS rDNA sequencing
The identity of the culture was checked with ITS rDNA sequencing. The axenic mycobiont cultures were used for DNA isolation using DNeasy Plant Mini Kit (Qiagen). Before the isolation procedure, the agar medium was mechanically separated from the culture and mainly the top part of the mycelium was used for DNA isolation. DNA was resuspended in sterile distilled water. PCR amplifications were performed using GeneAmp 9700 PCR Thermal Cycler (Applied Biosystems). One unit of RedTaq polymerase (Sigma) was used for each 50 µl of master mix containing 5 µl of 10× Taq polymerase reaction buffer, 0·2 mM of each of the four dNTP's and 0·5 µM of each primer. The primers ITS1F (Gardes & Bruns Reference Gardes and Bruns1993) and ITS4 (White et al. Reference White, Bruns, Lee, Taylor, Innis, Gelfand, Sninsky and White1990) were used for PCR and sequencing. The following thermal profile was employed: after an initial denaturation step at 95°C for 5 min, the PCR ran for 35 cycles (95°C for 1 min, 51°C for 40 s, 72°C for 1 min) with a final extension step at 72°C for 10 min. PCR products were resolved on 1% agarose gels in order to determine DNA fragment lengths, then purified using the High Pure PCR Product Purification Kit (Roche) and sequenced using the Macrogen (Korea) sequencing service (www.macrogen.com). The new ITS rDNA sequences from the mycobiont cultures of Protoparmeliopsis muralis were compared with the sequences available in GenBank using BLAST (Altschul et al. Reference Altschul, Gish, Miller, Myers and Lipman1990), in order to confirm their identity.
Microsatellite isolation
For DNA enrichment, we used the Fast Isolation by AFLP of Sequences Containing Repeats (FIASCO, Zane et al. Reference Zane, Bargelloni and Patarnello2002). This is based on the efficient digestion-ligation reaction of the amplified fragment length polymorphism (AFLP) procedure. DNA extracts obtained from mycobiont cultures were pooled and used for further experiments. Genomic fungal DNA was digested with the MseI enzyme and simultaneously ligated to MseI AFLP adaptors (5′-TAC TCA GGA CTC AT-3′/5′-GAC GAT GAG TCC TGA G-3′). The ligation-digestion mixture was then diluted and amplified with adaptor-specific primers MseI-N (5′-GAT GAG TCC TGA GTA AN-3′). The PCR reaction was optimized and the final thermal profile was the following: initial denaturation at 94°C for 2 min followed by 19 cycles of 94°C for 30 s, 53°C for 60 s and 72°C for 60 s, with a final elongation step at 72°C for 7 min. The resulting PCR product was used as a template for hybridization to biotynylated probes. We used the following biotynylated probes: (AG)10, (AC)10, (AT)10, (GC)10, (AAT)7, (AAC)7. DNA-probe hybrids were captured using Streptavidin-coated beads (Dynalbeads M-270 Streptavidin). The non-specific DNA was removed by 5 non-stringency (by adding 400 µl of TEN1000 buffer-10 mM Tris-HCl, 1 mM EDTA, 1 M NaCl, pH 7·5) and 5 stringency (by adding 400 µl of 0·2× SSC, 0·1% SDS) washes. DNA was separated from the beads-probe complex by two denaturation steps; first, by adding 50 µl of TE and incubating at 95°C for 5 min, after which the supernatant was removed and stored. In the second step, the beads were treated with NaOH. DNA was then precipitated and amplified using MseI-N primer. PCR products were used to produce a highly enriched microsatellite library.
Cloning of PCR products was carried out using the TOPO-TA Cloning Kit for sequencing (Invitrogen). Clones were tested for presence of inserts using a PCR screening test with M13Forward(-20) and M13Reverse primers. Plasmids containing different-sized PCR products were sequenced with T3 and T7 primers using Macrogen sequencing service.
Microsatellite identification and primer design
The sequences obtained were screened for the presence of repeats using RepeatMasker (Smit et al. Reference Smit, Hubley and Green1996–2010, http://www.repeatmasker.org/cgi-bin/WEBRepeatMasker), Perfect Microsatellite Repeat Finder (http://sgdp.iop.kcl.ac.uk/nikammar/repeatfinder.html) (Leach Reference Leach2000), and Microsatellite Repeats Finder (http://www.biophp.org/minitools/microsatellite_repeats_finder/demo.php) (Bikandi Reference Bikandi2006). The sequences from clones containing repeats of a sufficient length (at least 5 repeats) were chosen for primer design. The primers were designed using Primer3 software (Rozen & Skaletsky Reference Rozen, Skaletsky, Krawetz and Misener2000; http://frodo.wi.mit.edu/) and tested on mycobiont DNA extracts used for microsatellite isolation.
Primer test
The selected primers (Table 1) were tested on 21 specimens of Protoparmeliopsis muralis from several localities in Austria, the Czech Republic and Poland. Among the samples analyzed, ten specimens were collected from a single population in the Czech Republic (Rychtarov, Jihomoravský kraj). DNA was isolated according to the modified CTAB method (Guzow-Krzemińska & Węgrzyn Reference Guzow-Krzemińska and Węgrzyn2000) and PCR was performed using GeneAmp 9700 PCR Thermal Cycler (Applied Biosystems). One unit of RedTaq polymerase (Sigma) was used for each 50 µl of master mix containing 5 µl of 10× Taq polymerase reaction buffer, 0·2 mM of each of the four dNTP's and 0·5 µM of each primer (forward and reverse for each marker – Table 1). PCR amplification was confirmed on 2·5% agarose gels. In order to characterize the newly developed microsatellite markers, sequencing of PCR products was performed using Macrogen sequencing service. The new sequences for each marker were aligned using ClustalX software (Thompson et al. Reference Thompson, Gibson, Plewniak, Jeanmougin and Higgins1997) (with the following parameters: gap opening=15; gap extension=6·66). Sequences from different specimens which were identical were treated as a single allele and the unique multilocus genotypes were determined.
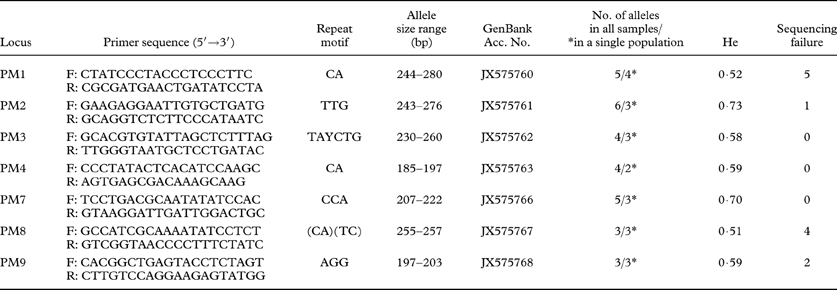
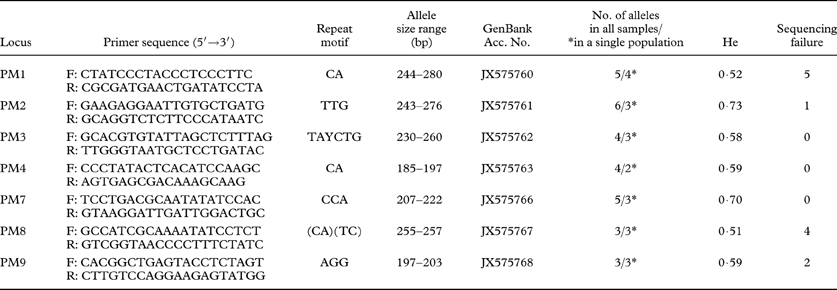
Table 1. Microsatellite primers developed in this study and characteristics of microsatellite loci that were analyzed in 21 individual samples of Protoparmeliopsis muralis. Primer names, sequence for each forward and reverse primers, repeat motifs, size range of the alleles (bp), GenBank accession numbers, number of alleles for all samples analyzed/and for 10 specimens selected from single population (marked with *), Nei's gene diversity (He) and number of samples for which amplification and sequencing failed.
Results and Discussion
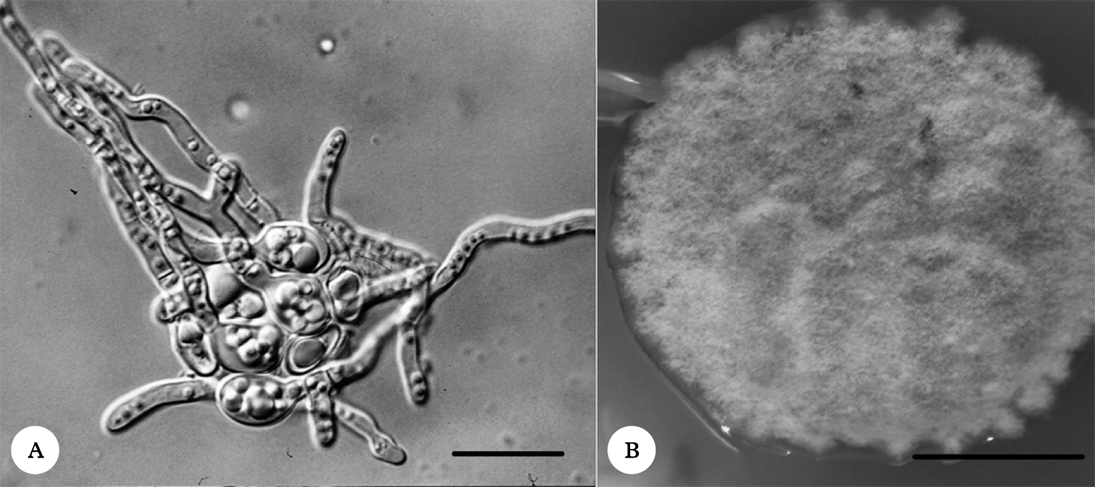
In order to avoid algal contamination, axenic mycobiont cultures were obtained from fungal spores (Fig. 1A). The multispore mycelia were subcultured on nutrient media (Fig. 1B) and the identities of mycelia were checked using ITS rDNA sequencing (GenBank Accession Numbers: KC791770 and KC791771) followed by BLAST analysis. The mycelia were subsequently used for microsatellite marker development according to the FIASCO procedure (Zane et al. Reference Zane, Bargelloni and Patarnello2002).
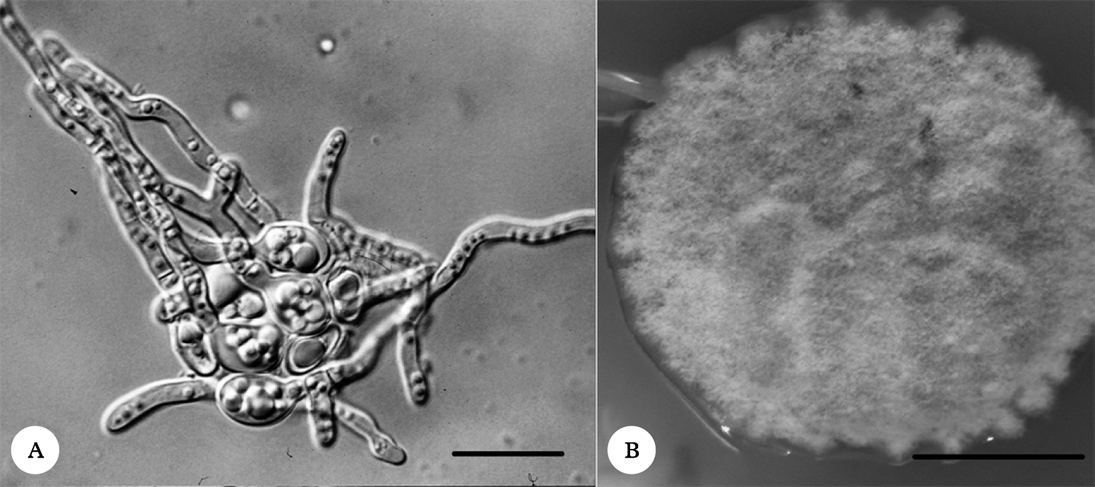
Fig. 1. A, germinating Protoparmeliopsis muralis spores; B, the mycelium of P. muralis grown on PDA medium. Scales: A=20 µm; B=1 cm.
In total, 380 clones were sequenced, of which 62 contained repeats. However, not all clones were unique; some of them were identical or chimeric sequences, which decreased the number of positive clones. Some of the positive clones were discarded due to either poor repeats or lack of a suitable sequence for primer design; for example, the microsatellite was located too close to the end of an insert and a flanking sequence was too short to design a primer, or the base composition was unsuitable.
Finally, 38 primer pairs were designed and tested. Among the primers tested some failed to amplify or produced multiple bands that were not interpretable. Many others were found to be monomorphic in the samples included in the analysis and therefore were excluded from further analyses.
Finally, 7 primer pairs (Table 1) were found to amplify polymorphic microsatellite loci from different specimens of Protoparmeliopsis muralis. The variability of some markers between specimens was documented on 2·5% agarose gels (Fig. 2). The sequences obtained were aligned and the number of repeats was determined for each locus. The number of alleles ranged from three to six per locus and Nei's gene diversity was calculated for each marker (Table 1).
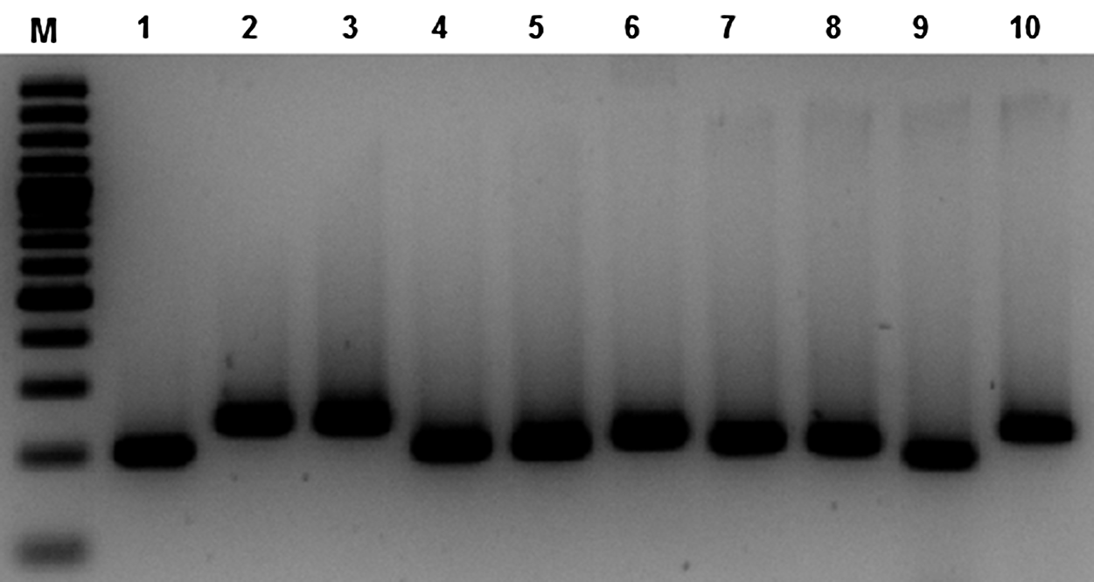
Fig. 2. The variation of PCR products of PM3 marker observed on 2·5% agarose gels. M, GeneRuler 100bp Plus DNA Ladder (Fermentas); 1–10, PCR products from specimens of Protoparmeliopsis muralis.
Not all loci yielded a PCR product from each sample. In some cases we failed to obtain the sequence of the marker due to poor amplification but no pattern was observed; the number of failures is given in Table 1. The best microsatellite markers that produced amplicons from all specimens were PM3, PM4 and PM7. On the other hand, with primers PM1 and PM8, 5 and 4 samples respectively were not amplified. However, the number of alleles determined for each locus does not necessarily correlate with the number of specimens analyzed; for example, for marker PM1, that revealed the most difficult amplification, we found five alleles. It might be possible that the most polymorphic markers fail to amplify more often because the flanking region also tends to be more variable. On the other hand, among 19 specimens analyzed, only 3 alleles of PM9 marker were identified. The variability of the markers for samples from a single population was equal (markers PM8 and PM9), or lower than the total number of alleles for all samples (Table 1).
We also defined unique multilocus genotypes within the samples analyzed. The missing data were treated as unknown and only genotypes that were different from others in at least one locus were defined as unique. Despite the missing data, we identified 19 different multilocus genotypes. Two specimens from a single population were found to be identical in all markers. Moreover, two other samples from different localities may in fact represent the same genotype although, due to the missing data from two markers, the determination of clonality cannot be made with absolute certainty. Out of ten samples from a single population, we identified nine different multilocus genotypes, showing that the markers reported here may be useful in further population studies of Protoparmeliopsis muralis.
Microsatellite markers are commonly used in population studies of plants and vertebrates due to their diversity. However, microsatellite loci seem to be less abundant in fungi than in other organisms (Dutech et al. Reference Dutech, Enjalbert, Fournier, Delmotte, Barrès, Carlier, Tharreau and Giraud2007). As summarized by Dutech et al. (Reference Dutech, Enjalbert, Fournier, Delmotte, Barrès, Carlier, Tharreau and Giraud2007), in many organisms the number of repeats was shown to be a good predictor of the level of variability. Also, Lim et al. (Reference Lim, Notley-McRobb, Lim and Carter2004), based on the analysis of 14 fungal genomes, showed that c. 90% of microsatellite loci had a low number of repeats (i.e. below eight); thus fungal microsatellites are expected to be less variable than in other taxa, mainly due to the low number of repeats. However, some mycobiont microsatellites have been shown to be more variable than photobiont loci (Dal Grande et al. Reference Dal Grande, Widmer, Wagner and Scheidegger2012; Werth & Scheidegger Reference Werth and Scheidegger2012). Although microsatellite markers could provide an excellent tool for population structure and gene flow studies, a very small number of lichens have been subjected to microsatellite development so far (i.e. Walser et al. Reference Walser, Sperisen, Soliva and Scheidegger2003; Magain et al. Reference Magain, Forrest, Sérusiaux and Goffinet2010; Jones et al. Reference Jones, Green, Hogg and Wilkins2012); thus more intensive studies are necessary for this group of organisms. Yet the symbiotic character of lichen-forming fungi is a disadvantage in the development of the new specific markers. A previous study by Widmer et al. (Reference Widmer, Dal Grande, Cornejo and Scheidegger2010) showed that five of twelve primer pairs developed for Lobaria pulmonaria by Walser et al. (Reference Walser, Sperisen, Soliva and Scheidegger2003) were algal-specific markers. However, the employment of axenic cultures for DNA isolation, further used in the marker development procedure, significantly decreases the risk of contamination. For lichens producing fruiting bodies, it could be advantageous to use multispore mycobiont cultures for the development of new markers.
Georg Brunauer, Martin Grube, Armin Hager, Martin Kukwa, Andreas Tribsch, Sabine Wornik, and Grzegorz Węgrzyn are acknowledged for valuable discussions. Two anonymous reviewers are thanked for their comments on the previous version of the manuscript. Hans Peter Comes is acknowledged for making his laboratory facilities available. The study was financially supported by the Marie Curie Fellowship within the 6th European Community Framework Programme, project no. 24206. BGK also acknowledges the support of the Marie Curie European Reintegration Grant within the 7th European Community Framework Programme, project no. 239343. EST-W gratefully acknowledges support from the Austrian Science Foundation Grant P23570.