


Kabuki syndrome was first described in Japanese children, in a pair of parallel reports in 1981,1, 2 with 20 cases reported in the medical literature by 1985.3 The syndrome, also known as Niikawa–Kuroki syndrome, is characterized by distinctive facial features, developmental delay, cardiac defects, and skeletal malformations.4 Its associated cardiovascular anomalies have been the subject of one detailed report,2 but have also been noted by other authors collecting series of cases.5, 6 A notable feature of many of the children having associated congenital cardiac disease is the late diagnosis of the syndrome, inferring a lack of recognition of the syndrome amongst paediatric cardiologists. We sought, therefore, to determine the prevalence of congenital cardiac malformations in, and the natural history of, children with Kabuki syndrome at our institution.
Methods
One of us (WR), a geneticist working in a paediatric environment, identified six children with Kabuki syndrome over a period of 1 year. The other (CM), a paediatric cardiologist, then reviewed the medical records, echocardiograms, surgical notes and genetic studies performed on each patient. The characteristic dysmorphic features were documented for each patient.
Results
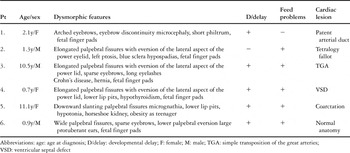
As indicated, six patients had been diagnosed with Kabuki syndrome during the period of study. There were three boys and three girls, with a median age of 1.7 years at diagnosis, and a range from 0.7 years to 11.1 years. For one patient, the mother was similarly affected. Although four of the cases were referred and diagnosed after the age of 2 years, two cases were not referred for diagnosis until after the age of 10 years, despite longstanding problems, with both children having required surgical intervention for congenital cardiac disease (Table 1). There were no cases of a second sibling affected. Significant developmental delay was evident in five patients, including delayed gross motor and cognitive functioning. Of note, five of the children had experienced significant problems with feeding, including either an aversion to food or refusal to eat, refractory gastroesophageal reflux, or oropharyngeal discoordination such as poor sucking and/or the ability to swallow, in the neonatal period. Indeed, one patient had required placement of a gastrostomy feeding tube.
Table 1. Characteristics of six children with Kabuki syndrome.
Dysmorphic features
All patients had some prominent findings in relation to the morphology of the eyebrows and eyelid, the typical profile observed being elongated palpebral fissures with eversion of the lateral aspect of the lower eyelid, giving an impression of ectropion (Fig. 1). Other findings included arched eyebrows, sometimes with sparse hair laterally, wide or slanting palpebral fissures, blue scleras, ptosis, absence of a segment of the eyebrow, and very prominent long eyelashes. Other facial abnormalities included microcephaly, a short philtrum, micrognathia, and large protuberant ears. In two patients, there was central pitting of the lower lip (Fig. 2). In four patients, there was prominence of the fetal finger pads (Fig. 3a,b). Finger pads on the volar surface of the digits are a normal embryological phenomenon, usually regressing by 15 weeks of gestation. Persistence of this clinical phenomenon is a valuable, nonspecific and easily overlooked clue to the diagnosis of Kabuki syndrome.
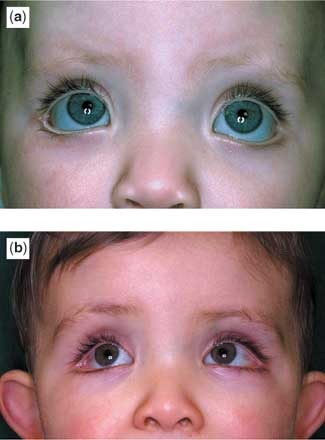
Figure 1. The (a, b) show the characteristic palpebral fissure of Kabuki syndrome. Note specifically the wide palpebral fissure, extending laterally and the visible blood vessels at the outer canthus of the eye. Additionally the lower eyelid shows an ectropion, which is unusual in a child but very characteristic in Kabuki syndrome. Note also the eyebrow, arched and with almost a discontinuity in the hairline in the middle of the eyebrow. As is typical of this syndrome, the eyelashes are long.
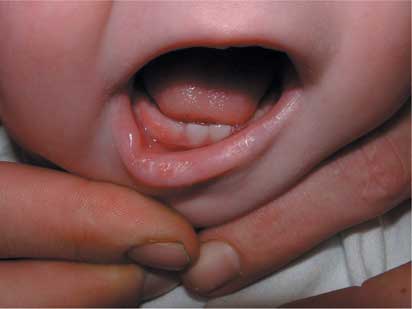
Figure 2. The pit in the lower lip.
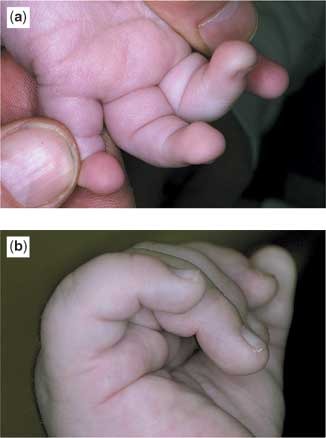
Figure 3. Fetal finger pads, characteristic for the syndrome, are shown from two different patients (a and b).
Congenital cardiac malformations
We found congenital cardiac lesions in five of the six patients (83% – Table 1). Surgical intervention was needed in three, including an arterial switch, construction of a Blalock–Taussig shunt, and repair of aortic coarctation. All patients had uneventful postoperative courses, and were discharged home within 1 week.
Involvement of other systems of organs
Additional problems included Crohn’s disease, hypothyroidism, and hypospadias, each seen in one patient, and inguinal hernias in two patients. Severe obesity developed in two patients once they became teenagers, despite an apparently normal caloric intake. All patients showed evidence of developmental delay. The routine karyotype was normal in these patients.
Discussion
Although Kabuki syndrome was first recognized in 1981, there has been a significant increase in interest in the syndrome in recent years.1–8 The diagnosis is primarily based upon the recognition of the characteristic dysmorphic facial features, in association with at least three additional features from the list of developmental delay, congenital cardiac defects, short stature, skeletal anomalies, and fetal finger pads.4 The dysmorphic features typically involve the periocular region. Specifically, there can be ptosis, elongated palpebral fissures, tilted optic discs which does not affect vision, everted lower lateral eyelids, arched eyebrows and interrupted eyebrows. All these features are well described, and well recognized as cardinal features.1–7 Other clinical features may include hypotonia, joint laxity, cleft palate in up to half, hypodontia, a full lower lip with either nodules or lip pits, a prominent nuchal fold during fetal life, and neonatal hydrops.7 The onset of severe obesity, as noted in two of our patients, both on entering adolescence, is also well established, having been highlighted by Schrander-Stumpel et al. in their review of 29 cases.7
The aetiology of the syndrome remains unknown, with the risk of familial recurrence estimated at approximately 2%. Although one group claimed to have shown duplication of chromosome 8p22–8p23.1 by comparative genomic hybridization and bacterial artificial chromosomes fluorescent in-situ hybridization,9 this finding has been questioned by others.10, 11 Given the similarity between the cardiac defects observed in Kabuki and Turner syndromes, overexpression of X chromosome-derived sequences have also been proposed to be associated with the Kabuki-like phenotype.10, 12 Evidence for involvement of the X chromosome as a causal agent, however, is lacking, and an objective laboratory-based confirmatory test for the syndrome is not yet established. There have been instances of likely transmission from parent to child, which has raised speculation that the syndrome is caused by a tiny submicroscopic deletion on an unidentified autosome.8, 13
Involvement of multiple systems of organs has previously been described, including epilepsy with and without polymicrogyric changes,14, 15 hepatic involvement, including fibrosis and sclerosing cholangitis,16 autoimmune disorders, malignancies such as leukaemia,17 and endocrinopathies, including diabetes and deficiency of growth hormone.18 The finding of associated Crohn's disease in one of our patients has not, to the best of our knowledge, been previously reported.
Our experience serves further to highlight the high prevalence of congenital cardiac malformations in the setting of Kabuki syndrome, primarily those involving the ventricular outlets, the aortic arch, and septal defects. Digilio et al.4 reported cardiac malformations in three-fifths of their cohort of 60 patients, with two-thirds of those with cardiac defects being male. This compared to a prevalence of four-fifths in our significantly smaller group of patients. The most common defects in the cohort collected by Digilio et al. were coarctation, with or without other left-sided obstructive lesions, atrial septal defect, and ventricular septal defect, each lesion seen in about one-fifth of the cohort. Less common cardiac defects included aortic stenosis, pulmonary stenosis, tetralogy of Fallot, double outlet right ventricle, complete transposition, and Ebstein’s malformation. In their study, standard chromosomal analysis and examination for 22q11 deletion using fluorescent situ hybridization revealed abnormalities in four patients. An inherited balanced (3; 10) (p25; p15) translocation was found in an affected brother and sister, and a fragile site at 16q22.1 was detected in two patients. Microdeletion of 22q11 was excluded in all patients, and has never been described in the setting of Kabuki syndrome. As in the general population, karyotypes were normal in our children. In the absence presently of a more specific diagnostic genetic test, the diagnosis remains clinical, based on the characteristics facial features and other components outlined above. Although the syndrome can be difficult to diagnose in the first year of life, requiring a practiced eye, two of our patients were not diagnosed until after the 10th year of age, highlighting that subtle features of this syndrome may go unrecognized for some time. The cohort examined by Digilio et al.4 included a wide range of patients, with mean age 8.3 plus or minus 4.0 years, but they did not comment on the timing of diagnosis.
In conclusion, paediatric cardiologists should be alert to the possibility of Kabuki syndrome in any child with a history of developmental delay, severe feeding problems in the neonatal period disproportionate to the degree of cardiac disease, the presence of a cardiac defect, and the characteristic dysmorphic findings as described herein. Earlier detection of this syndrome may have important implications for the long-term prognosis of such children.




