


Serious environmental problems including global warming and local pollution are associated directly with excessive usage of fossil fuels (Fayaz et al., Reference Fayaz, Saidur, Razali, Anuar, Saleman and Islam2012; Xie et al., Reference Xie, Peng, Peng, Gu and Liu2014). ‘Green’ energy to replace fossil fuels has become most challenging energy issue of the last few decades (Roszak et al., Reference Roszak, Firlej, Roszak, Pfeifer and Kuchta2016; Shervani et al., Reference Shervani, Mukherjee, Gupta, Mishra, Illath, Ajithkumar, Sivakumar, Sen, Balani and Subramaniam2017). Hydrogen is a promising candidate as an energy source due to its high energy density per unit mass and availability; because hydrogen can be produced from renewable resources via photoelectrochemical and biological processes, it has minimal environmental impact (Zhang et al., Reference Zhang, Zhang, Zhang, Wang and Yang2014; Ren et al., Reference Ren, Musyoka, Langmi, Mathe and Liao2017). Storage is an important step in the utilization of hydrogen as a green fuel (Mondelli et al., Reference Mondelli, Bardelli, Vitillo, Didier, Brendle, Cavicchia, Robinet and Charlet2015). The potential for safe and affordable storage of hydrogen in materials has attracted considerable attention recently (Hörtz et al., Reference Hörtz, Ruff and Schäfer2015; Alver, Reference Alver2017; Wei et al., Reference Wei, Lin, Tseng and Chan2017). Among the various storage methods, adsorption is one of the most popular because it is simple to use (Niaz et al., Reference Niaz, Manzoor and Pandith2015). Therefore, many investigators have studied the storage of H2 experimentally using chemical and physical adsorption in different materials, including metals (Sun et al., Reference Sun, Yang, Yang, Fan, Peng, Long, Zhou, Zu and Du2016; Bachurin & Viadimirov, Reference Bachurin and Viadimirov2017), alloys (Mahdi & Sahar, Reference Mahdi and Sahar2015), minerals (Arean et al., Reference Areán, Palomino, Carayol, Pulido, Rubeš, Bludský and Nachtigall2009; Henkel et al., Reference Henkel, Pudlo, Werner, Enzmann, Reitenbach, Albrecht, Würdemann, Heister, Ganzer and Gaupp2014; Sigot et al., Reference Sigot, Ducom and Germain2016) and organometallic compounds (Gu et al., Reference Gu, Gao and Yu2004). Concerning sorbent materials, natural or modified clay minerals have received much attention as possible low-cost and high-thermal-stability adsorbents in the storage of H2 removed from contaminated air (Itadania et al., Reference Itadania, Tanaka, Abe, Taguchi and Nagao2007; Charlet et al., Reference Charlet, Alt-Epping, Wersin and Gilbert2017). Kaolinite is one of the most abundant clay minerals (Brigatti et al., Reference Brigatti, Galan, Theng, Bergaya, Theng and Lagaly2006; Chen & Lu, Reference Chen and Lu2015) and is very common in soils, especially in tropical and sub-tropical areas. The storage of H2 on kaolin clays is based on an adsorption mechanism. The use of kaolinite to adsorb gases such as hydrogen, water and carbon dioxide has been reported in the past (Venaruzzo et al., Reference Venaruzzo, Volzone, Rueda and Ortida2002; Saada et al., Reference Saada, Gaboriau, Cornu, Bardot, Villieras and Croue2003). Cations on kaolinite surfaces create a strong electric field that favours gas adsorption. So far, experimental methods of adsorption usually provide enthalpies and sometimes equilibrium constants, but they do not provide information about the geometry of adsorption on the sorbent. A theoretical analysis of the adsorption mechanism of H2 monomers on natural kaolinite from a microscopic point of view would improve understanding of the adsorptive properties of the kaolinite–H2 interface and the influence of H2 adsorbed on clay minerals. Computational chemistry calculations based on density functional theory (DFT) have proven to be a powerful and reliable tool to study H2–solid interfaces at the microscopic level. Hence, a greater insight into the process of H2 adsorption on the kaolinite (001) surface through detailed first-principles analysis is necessary.
Existing experimental data (Adams, Reference Adams1983; Bish, Reference Bish1993; Benco et al., Reference Benco, Tunega, Hafner and Lischka2001), calculated results (Hess & Saunders, Reference Hess and Saunders1992; Hayashi, Reference Hayashi1997; Hobbs et al., Reference Hobbs, Cygan, Nagy, Schultz and Sears1997; Plançon & Giese, Reference Plançon, Giese, Snyder, Drits and Bookin1997; Teppen et al., Reference Teppen, Rasmussen, Bertsch, Miller and Schäferll1997; Hu & Michaelides, Reference Hu and Michaelides2008) and data on the kaolinite layers with the ideal structural formula Al2Si2O5(OH)4 are based on the 1:1 layer structure, consisting of a tetrahedral (SiO4) sheet in which Si atoms are coordinated by oxygen anions and an octahedral (AlO6) sheet where Al atoms are coordinated by oxygen atoms and hydroxyl groups (Bailey, Reference Bailey, Brindley and Brown1980). Quantitative estimates indicate that there is a certain degree of van der Waals attraction and hydrogen bonding between the silicate (SiO4) sheet and the adjoining aluminate (AlO6) sheets (Sato et al., Reference Sato, Ono, Johnston and Yamagishi2005; Hajjaji et al., Reference Hajjaji, Andrejkovicova, Pullar, Tobaldi, Lopez-Galindo, Jammousi, Rocha and Labrincha2016). Kaolinite microparticles exist as hexagonal plates with dominant (001) basal surfaces that are almost perfectly cleaved; this is the plane that is mainly exposed in kaolinite crystals (Giese, Reference Giese1973; Šolc et al., Reference Šolc, Gerzabek, Lischka and Tunega2011). As reported by Zhang et al. (Reference Zhang, Zhang, Zhang, Wang and Yang2014), the hydroxyl groups of the Al–O surface are supposed to form hydrogen bonds with molecules such as water and carbon dioxide (Hu & Michaelides, Reference Hu and Michaelides2008; He et al., Reference He, Zhao and Li2014). Thus, the hydroxyl (001) surface is the surface of primary interest in adsorption studies. Calculations were also performed to determine the adsorption energy of hydrogen molecules on the tetrahedral (00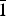 $ {\bar 1} $) and hydroxylated (001) surfaces of kaolinite. The results showed that a hydrogen molecule adsorbs more weakly on the tetrahedral surface than it does on the octahedral surface. The objectives of this study were to investigate H2 adsorption sites, adsorption energies, charge transfer, H2 structure during adsorption and the structure of the intermediate reaction complex.
$ {\bar 1} $) and hydroxylated (001) surfaces of kaolinite. The results showed that a hydrogen molecule adsorbs more weakly on the tetrahedral surface than it does on the octahedral surface. The objectives of this study were to investigate H2 adsorption sites, adsorption energies, charge transfer, H2 structure during adsorption and the structure of the intermediate reaction complex.
METHOD OF CALCULATIONS
Periodic DFT calculations were performed using the frozen-core all electron projector augmented wave method in the Vienna ab initio simulation package (VASP) (Kress & Furthmüller, Reference Kresse and Furthmüller1996). The Kohn–Sham DFT equations were solved using plane-wave pseudopotentials and periodic boundary conditions. The local density approximation of electron exchange potential and correlation energy was used. The electron–ion interaction is described by Blöchl's projector augmented wave method, which takes the exact shape of the valence wave functions into account (Blöchl, Reference Blöchl1994; Kresse & Joubert, Reference Kresse and Joubert1999). The converged kinetic energy cut-off was set to 400 eV, which was sufficient to ensure that the error is <0.01 eV in the calculated values for energies and <0.001 Å for the primitive bulk cell. Monkhorst–Pack meshes (Monkhorst & Pack, Reference Monkhorst and Pack1976) of a 3 × 3 × 1 k-point grid in the Brillouin surface for the p (2 × 2) surface cell were used.
The kaolinite (001) surface was modelled using a slab composed of ‘H–O–Al–O–Si–O’ six atomic sublayers with a vacuum thickness of 20 Å. Based on the data of Hess and Saunders (Reference Hess and Saunders1992), the calculated lattice parameters of bulk kaolinite were a = 5.155 Å, b = 5.155 Å, c = 7.405 Å, α = 75.14°, β = 84.12° and γ = 60.18°, and these were used in the present study. Adsorbates were placed on one side of the slab and a dipole correction was included for all slab calculations. During the calculation, all the H, O and Al atoms in the three sublayers (AlO6 surface), as well as the H2, were allowed to relax while the other three atomic sublayers (SiO4 surface) of the slab were kept fixed at the calculated bulk positions. In the present study, calculations for adsorbed H2 molecules at surface coverages that ranged from 1/16 to 1 monolayer (ML) were performed for nine adsorption sites. With regards to the inner-surface hydroxyl groups, the nine adsorption sites included three one-fold top sites (T1–T3), two two-fold bridge sites (B1–B2) and four three-fold cavity sites (H1–H4). Figure 1 shows the hydroxylated (001) surface of kaolinite after relaxation, in which two-thirds of the surface hydroxyl groups tilt (T1–T2) and the other third of hydroxyl groups are almost parallel (T3) to the surface. The adsorption of H2 on the cavity sites for 1/16, 1/8, 1/4, 1/2, 3/4 and 1 ML with the p (2 × 2) surface cell and the coverage of 1/8, 1/4, 1/2, 3/4 and 1 ML of H2 molecules on the top and bridge sites were calculated systematically, respectively. Several sizes of the kaolinite (001) model were used to test the influence of model size on H2 adsorption energy.

Fig. 1. Top view of kaolinite (001) surface with (a) three top adsorption sites (T1–T3), (b) two bridge adsorption sites (B1–B2) and (c) four cavity adsorption sites (H1–H4).
RESULTS AND DISCUSSION
In the present study, the adsorption energy (E ads) is the average adsorption energy of the H2 molecules on kaolinite substrate, defined as
 $$ \eqalign{& E_{{\rm ads}}(\Theta ) =\cr & - \displaystyle{\hbox{1} \over {N_{{\rm H}_2}}} \lsqb E_{{\rm H}_2{\rm /kaolinite(001)}} - E_{{\rm kaolinite(001)}} - N_{H_2}E_{H_2} \rsqb} $$
$$ \eqalign{& E_{{\rm ads}}(\Theta ) =\cr & - \displaystyle{\hbox{1} \over {N_{{\rm H}_2}}} \lsqb E_{{\rm H}_2{\rm /kaolinite(001)}} - E_{{\rm kaolinite(001)}} - N_{H_2}E_{H_2} \rsqb} $$where 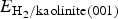 $E_{{\rm H}_2{\rm /kaolinite (001)}}$ and E kaolinite (001) are total energies of an N hydrogen adsorption system and the corresponding clean kaolinite surface, respectively.
$E_{{\rm H}_2{\rm /kaolinite (001)}}$ and E kaolinite (001) are total energies of an N hydrogen adsorption system and the corresponding clean kaolinite surface, respectively. 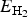 $E_{{\rm H}_2}$ is the total energy of a free hydrogen molecule,
$E_{{\rm H}_2}$ is the total energy of a free hydrogen molecule, 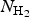 $N_{{\rm H}_2}$ is the total number of hydrogen molecules adsorbed and Θ is defined as the ratio of the number of H2 molecules adsorbed to the total number of molecules adsorbed on the corresponding top, bridge or cavity sites in an ideal kaolinite (001) surface. With this definition, a positive value of the adsorption energy indicates that the adsorption is an exothermic (stable) process and a negative value indicates an endothermic (unstable) reaction. Similar to Hu and Michaelides (Reference Hu and Michaelides2008), all of the three kinds of high-symmetry adsorption sites on the (001) surface were considered. Two original molecular configurations of upright and recumbent CH4 with respect to the surface were examined at all adsorption sites. After optimizing the adsorption models, the three adsorption states of the top (T1–T3), bridge (B1–B2) and cavity sites (H1–H4) with tilted (Figs 2a, c, d) or perpendicular (Fig. 2b) H2 molecules were stabilized. The H−H bond of adsorbed H2 molecules on the top (T3) adsorption sites is perpendicular to the surface (named as top-z for clarity) after relaxation, and the remainder form acute angles with the surface. The perpendicular and tilted orientation represented configurations in which one hydrogen atom of H2 formed a bond with the surface. All top (T1–T2), bridge (B1–B2) and cavity (H1–H4) adsorption sites for H2 molecules had similar adsorption energies in the coverage regime of 0 < Θ ≤ 1 ML, respectively. The calculated adsorption energies (E ads) of H2 on these four types of surface sites with respect to the free molecule H2 are summarized and illustrated for different H2 coverages for 0 < Θ ≤ 1 ML (Fig. 3; Table 1).
$N_{{\rm H}_2}$ is the total number of hydrogen molecules adsorbed and Θ is defined as the ratio of the number of H2 molecules adsorbed to the total number of molecules adsorbed on the corresponding top, bridge or cavity sites in an ideal kaolinite (001) surface. With this definition, a positive value of the adsorption energy indicates that the adsorption is an exothermic (stable) process and a negative value indicates an endothermic (unstable) reaction. Similar to Hu and Michaelides (Reference Hu and Michaelides2008), all of the three kinds of high-symmetry adsorption sites on the (001) surface were considered. Two original molecular configurations of upright and recumbent CH4 with respect to the surface were examined at all adsorption sites. After optimizing the adsorption models, the three adsorption states of the top (T1–T3), bridge (B1–B2) and cavity sites (H1–H4) with tilted (Figs 2a, c, d) or perpendicular (Fig. 2b) H2 molecules were stabilized. The H−H bond of adsorbed H2 molecules on the top (T3) adsorption sites is perpendicular to the surface (named as top-z for clarity) after relaxation, and the remainder form acute angles with the surface. The perpendicular and tilted orientation represented configurations in which one hydrogen atom of H2 formed a bond with the surface. All top (T1–T2), bridge (B1–B2) and cavity (H1–H4) adsorption sites for H2 molecules had similar adsorption energies in the coverage regime of 0 < Θ ≤ 1 ML, respectively. The calculated adsorption energies (E ads) of H2 on these four types of surface sites with respect to the free molecule H2 are summarized and illustrated for different H2 coverages for 0 < Θ ≤ 1 ML (Fig. 3; Table 1).

Fig. 2. Top view of H2 molecule adsorbed on the (a) top, (b) top-z, (c) bridge and (d) cavity sites of kaolinite (001) surface.

Fig. 3. Calculated adsorption energy (E ads) of the H2/kaolinite (001) surface vs. the coverage for the H2 molecule adsorption in various sites. The solid lines connecting the calculated adsorption energies are used as visual guides. ML = monolayer.
Table 1. The calculated adsorption energy (E ads, eV), as a function of molecular H2 coverage on the various sites of kaolinite (001).

ML = monolayer.
A tilted H2 on the cavity site at a coverage of 1/16 ML was energetically stable, followed in order of reducing stability by tilted H2 on the bridge and the top (T1–T2) sites at a coverage of 1/8 ML. Here, the adsorption energies of H2 on the kaolinite (001) surface were 0.17, 0.28 and 0.39 eV for the top (T1–T2), bridge and cavity sites, respectively. The adsorption energy of the H2 molecule on the T3 (top-z) site was 0.22 eV at Θ = 1/4 ML, which was higher than the top (T1–T2) sites but lower than the bridge and cavity sites. At the highest coverage of 1 ML, an inclined H2 molecule was preferably adsorbed on the cavity site, and the following stable adsorption sites were the bridge, top-z and top sites. The adsorption energies of H2 on the kaolinite (001) surface were 0.14, 0.18, 0.20 and 0.26 eV for the top, top-z, bridge and cavity sites, respectively. The calculated adsorption energies of H2 (Fig. 3) revealed that the cavity site was more stable than the bridge, top-z and top sites in the coverage regime of 0 < Θ ≤ 1 ML. Meanwhile, the quantities of the top, top-z, bridge and cavity adsorptions displayed a modestly decreasing tendency with the increase in H2 adsorption, while the overall variation of the magnitude of E ads was rather small in the range of coverage. The decrease in adsorption with coverage indicated lower stability of surface adsorption due to the repulsion of neighbouring H2 molecules.
Calculated geometries for H2 molecule adsorption on the top, top-z, bridge and cavity sites of kaolinite (001) at Θ = 1/4, 1/2, 3/4 and 1 ML, including the H–H bond lengths d H–H (Å), the angle of the H–H bond with the surface (∠HHS) in degrees and the height  $h_{{\rm H}_2 - {\rm H}}$ of adsorbate H2 above the (001) surface, are summarized in Table 2. For all the adsorption sites, the H–H bond lengths d H–H of the H2 molecule increased slightly from 0.75 Å in the gas phase (Ganji et al., Reference Ganji, Sharifi, Ahangari and Khosravi2014; Yu et al., Reference Yu, Liu, Wang, Jia, Hou, Si, Li and Zhao2018) to 0.78 Å with increasing Θ values. Furthermore, H2 was adsorbed on the top, bridge and cavity sites with tilt angles of 17.8°, 47.1° and 55.3°, respectively, at Θ = 0.25 ML. The angles on these adsorption sites decreased with increasing coverage. By contrast, the H–H bonds of H2 molecules adsorbed on the top-z sites were almost perpendicular to the surface with angles of ~90° for coverage 0 < Θ ≤ 1 ML. With respect to the height
$h_{{\rm H}_2 - {\rm H}}$ of adsorbate H2 above the (001) surface, are summarized in Table 2. For all the adsorption sites, the H–H bond lengths d H–H of the H2 molecule increased slightly from 0.75 Å in the gas phase (Ganji et al., Reference Ganji, Sharifi, Ahangari and Khosravi2014; Yu et al., Reference Yu, Liu, Wang, Jia, Hou, Si, Li and Zhao2018) to 0.78 Å with increasing Θ values. Furthermore, H2 was adsorbed on the top, bridge and cavity sites with tilt angles of 17.8°, 47.1° and 55.3°, respectively, at Θ = 0.25 ML. The angles on these adsorption sites decreased with increasing coverage. By contrast, the H–H bonds of H2 molecules adsorbed on the top-z sites were almost perpendicular to the surface with angles of ~90° for coverage 0 < Θ ≤ 1 ML. With respect to the height 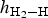 $h_{{\rm H}_2 - {\rm H}}$ of adsorbate H2 above the kaolinite surface, for the cavity adsorption site, the value of
$h_{{\rm H}_2 - {\rm H}}$ of adsorbate H2 above the kaolinite surface, for the cavity adsorption site, the value of 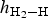 $h_{{\rm H}_2 - {\rm H}}$ was slightly smaller than that for the top, top-z and bridge sites at the coverage range of 1/4 ≤ Θ ≤ 1 ML (Table 2). A short height
$h_{{\rm H}_2 - {\rm H}}$ was slightly smaller than that for the top, top-z and bridge sites at the coverage range of 1/4 ≤ Θ ≤ 1 ML (Table 2). A short height 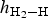 $h_{{\rm H}_2 - {\rm H}}$ implied a strong interaction between H2 and the kaolinite surface. Also, the cavity sites were the most stable. The calculated adsorbate height of H2 on these four types of surface sites are illustrated for different coverages in the regime 1/4 ≤ Θ ≤ 1 ML (Fig. 4). Note that for all four types of adsorption sites, the values of
$h_{{\rm H}_2 - {\rm H}}$ implied a strong interaction between H2 and the kaolinite surface. Also, the cavity sites were the most stable. The calculated adsorbate height of H2 on these four types of surface sites are illustrated for different coverages in the regime 1/4 ≤ Θ ≤ 1 ML (Fig. 4). Note that for all four types of adsorption sites, the values of 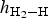 $h_{{\rm H}_2 - {\rm H}}$ increased with increasing Θ, which was consistent with the fact that the stability of adsorbed H2 decreased with increasing coverage.
$h_{{\rm H}_2 - {\rm H}}$ increased with increasing Θ, which was consistent with the fact that the stability of adsorbed H2 decreased with increasing coverage.

Fig. 4. Calculated adsorbate height, 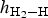 $h_{{\rm H}_2 - {\rm H}}$, of H2 above the surface vs. the coverage for the H2 molecule adsorption on various sites. The solid lines connecting the calculated heights are used as visual guides. ML = monolayer.
$h_{{\rm H}_2 - {\rm H}}$, of H2 above the surface vs. the coverage for the H2 molecule adsorption on various sites. The solid lines connecting the calculated heights are used as visual guides. ML = monolayer.
Table 2. The calculated adsorbate heights (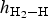 $h_{{\rm H}_2 - {\rm H}}$), angles of the H–H bond with the surface (∠HHS) and H–H bond lengths (d H-H) for various coverages of atomic H2 adsorption on the kaolinite (001) surface.
$h_{{\rm H}_2 - {\rm H}}$), angles of the H–H bond with the surface (∠HHS) and H–H bond lengths (d H-H) for various coverages of atomic H2 adsorption on the kaolinite (001) surface.

During geometry optimization, the distances between the interlayer of the outermost three atomic layers of kaolinite were changed. The changes of Δd ij were calculated according to the equation Δd ij = (d ij – d 0) / d 0, where d ij and d 0 are the distance between the ith and jth layers of the relaxed surface and the corresponding distance between the ith and jth layer of the clean kaolinite along the (001) direction, respectively. The calculated relaxations for the kaolinite (001) surface are summarized in Table 3. The calculated results showed that the adsorption of H2 on kaolinite (001) induced notable changes in the interlayer distance of the substrate. After adsorption of H2 molecules at the top and top-z sites, the distance between the first and second layer, Δd 12, was positive from 0.39% to 0.57% and from 0.83% to 1.88%, but the Δd 23 decreased from 0.16% to 0.13% and from 0.11% to 0.06%, respectively, for coverage 0 < Θ ≤ 1 ML. Therefore, the distance between the topmost two atomic layers expanded, but the distance between the second and third layers of the kaolinite (001) surface contracted with increasing H2 coverage. Analogously, for the bridge and cavity sites, the Δd 12 was negative from –3.44% to –1.92% and –2.38% to –0.80%, and the Δd 23 increased from –0.31% to –0.13% and –0.06% to 0.01% with increasing H2 coverage, respectively. These changes reflected the strong influence of the H2 adsorbates on the neighbouring H and O atoms and, thus, resulted from significant redistribution of the electronic structure. H2 adsorption caused the outermost kaolinite (001) layer separation to relax back to something close to its ‘ideal’ bulk value.
Table 3. The calculated interlayer relaxations (Δd 12 and Δd 23) for various coverages of atomic H2 adsorption on the kaolinite (001) surface.

ML = monolayer.
To gain more insights into the precise nature of the chemisorbed molecular state in the H2/kaolinite (001) system, the electronic partial density of state (PDOS) of the H2 molecule and the neighbouring H and O atoms of the (001) surface were calculated. The results were analysed by means of the electron density difference Δρ(r), which was obtained by subtracting the electron densities of non-interacting component systems, ρ kaolinite(001)(r) + ρH 2(r), from the density ρ(r) of the H2/kaolinite (001) system, while retaining the atomic positions of the component system at the same location as in H2/kaolinite (001). Positive (dark) Δρ(r) values indicated an accumulation of electron density upon binding, while negative (grey) values corresponded to electron density depletion.
As a typical example, the PDOSs of the adsorbed H2 orbitals (s and p) and the substrate H and O atoms coordinated with H2 on the two stable adsorption configurations of the top (T1–T2) and cavity sites were plotted (Fig. 5); the electron density differences are shown in Figs 5a and c (insets). For comparison, the PDOSs of the free H2 molecule and the corresponding neighbouring O and H atoms of clean kaolinite (001) surface were also calculated. After adsorption of the H2 molecule on the top site of kaolinite (001), both the s and p orbitals of H2 shifted down in energy by ~4.9 eV. Furthermore, the amplitudes of sp bonding orbitals were weaker than those in the free H2. By contrast, the sp orbitals of the surface H and O atoms had a small shift upwards with respect to the Fermi level. As the sp orbitals of adsorbed H2 aligned with the p bonding orbital of the adsorbed neighbouring O atom of kaolinite (001), the energy ranged from –6.72 to –4.77 eV (Figs 5a, b). These features were essentially caused by the different electronegativities of kaolinite and H2 molecules, which induced charge redistribution and thus built a global electrostatic attraction between the H2 molecule and neighbouring H and O atoms. The result was substantiated by the 3D electron density difference (inset of Fig. 5a). A remarkable charge accumulation existed between the adsorbate and substrate and an H−H bond was formed.

Fig. 5. The partial density of state plots for the H2 molecule and the neighbouring O and H atoms bonded to H2 at the stable top and cavity adsorption sites on the surface: (a) free and adsorbed H2 molecule at the top adsorption site; (b) clean and adsorbed kaolinite (001) surface at the top adsorption site; (c) free and adsorbed H2 molecule at the cavity adsorption site; (d) clean and adsorbed kaolinite (001) surface at the cavity adsorption site. The insets show the side views of electron density differences for the H2 atoms at the stable (a) top and (c) cavity adsorption sites. The Fermi energy is set at zero.
The PDOS of the tilted H2 adsorbed on a cavity site is depicted in Fig. 5c and d. A new peak at –5.76 eV below the Fermi level was observed in the PDOS. This new peak was contributed by the s hybridization with kaolinite s and p orbitals. The orbitals of the H2 molecule were shifted to lower energy and the amplitudes of the sp orbitals were weaker than those in a free H2 molecule, even in the H2 adsorbed on the top site. Furthermore, the overlap between adsorbed H2 and neighbouring O and H atoms of kaolinite (001) surface electrons in the energy ranged from –6.69 to –3.27 eV. The 3D electron density difference distribution of H2 was calculated (Fig. 5c, inset), which revealed the redistribution of charge after adsorption. For adsorption in cavity sites, the charge depletion was mainly distributed around the H atoms of the kaolinite (001) surface, while a manifested charge accumulation was observed in the H atoms of H2, displaying acceptance of electrons from the sp states of surface H and O atoms. The above results illustrated that the cavity was more stable than the top adsorption site for H2 molecules.
The orbital-resolved PDOS for the H2 adsorption on the bridge site and the neighbouring O and H atoms at Θ = 1/8 and Θ = 1 ML are shown in Fig. 6a and b, respectively. At a high coverage (Θ = 1 ML), the narrow amplitude peak at − 5.37 eV denoted the ‘H2 s state’ (Fig. 6b), which was mainly hybridized with the sp state of the neighbouring H and O atoms of the (001) surface. At a low coverage (Θ = 1/8 ML), the amplitudes of the sp orbitals of H2 molecules were much weaker than those in the case of Θ = 1 ML. Furthermore, compared to Θ = 1 ML, the hybridization of H2 s and surface H and O sp states was distinctly enhanced in the case of Θ = 1/8 ML. In particular, the main peak at E = ~−4.59 eV in the H2 s PDOS (Fig. 6a) resulted from the hybridization between adsorbate and substrate. The new H2 peak at − 6.01 eV below the Fermi level observed in Fig. 6a showed that the peak was contributed by the s hybridization with kaolinite s and p orbitals. These results revealed that the s orbital hybridized strongly with kaolinite sp orbitals and that the covalent H–H bond between H2 molecules and the kaolinite (001) surface decreased with the increasing coverage of H2 molecules.

Fig. 6. The partial density of state plots for the bridge adsorption site on a surface H2 molecule and the neighbouring H and O atoms at (a) Θ = 1/8 and (b) Θ = 1 ML, respectively. The Fermi level is set at zero. ML = monolayer.
CONCLUSIONS
The first-principles total energy calculation was used to investigate systematically the mechanism of H2 molecule adsorption on the kaolinite (001) surface, the adsorption energy and the changes in atomic and electronic structures. Different adsorption sites have been considered using surface models (p [2 × 2] surface unit cells) in a wide range of coverage from 1/16 to 1 ML. The three-fold cavity site is the most stable among all possible pure adsorbtion sites, followed by the bridge, top-z and top sites. Adsorption energy at the most stable position was obviously greater than at the other adsorption sites at the same coverage. Remarkably, this influence on the energy decreased with increasing H2 coverage. The decrease in the H2 adsorption energy for all four types of sites in the coverage range 0 < Θ ≤ 1 implied the lower stability of surface adsorption due to the repulsion of neighbouring H2 molecules. Different coverage has obvious effects on the atomic geometry, the charge density distribution and the electronic structure of the adsorbed H2 on the kaolinite (001) surface. The H–H bond lengths d H–H of the H2 molecule on all adsorption sites increased slightly from 0.75 Å in the gas phase to 0.78 Å with increasing Θ values. The distances between the interlayers of the three outermost atomic layers of kaolinite changed significantly, which underlined the fundamental influence of covalent bonding between the H2 molecule and kaolinite surface H atoms. Furthermore, the changes in the orbital-resolved PDOS and the distribution of electron density difference of H2 and O and H atoms of the surface were smaller with increasing coverage, which indicated that the hydrogen molecules are easily adsorbed with lower coverage. The above results will be helpful for future theoretical studies of the adsorption behaviour of H2 on the kaolinite (001) surface, which is of key importance in H2 storage.
ACKNOWLEDGMENTS
This research was supported by the National Key Research and Development Program (No. 2016YFC060090X), the Program for the National Natural Science Foundation of China (No. 41702317 and 51574296) and the Young Elite Scientist Sponsorship Program by the Chinese Association for Science and Technology (CAST).









