INTRODUCTION
Strains of Trypanosoma cruzi, the agent of Chagas disease, which is widely distributed in Latin America, shows considerable biological differences in growth rate, histotropism, pathogenicity, infectivity for vectors and mammal hosts and drug susceptibility. Moreover, Chagas disease is characterized by a wide spectrum of clinical manifestations that can occur during the acute and chronic phases. Some evidence suggests that parasite factors can explain this variability. In attempts to find the basis of this heterogeneity, a variety of genetic typing was developed. Multilocus enzyme electrophoresis (MLEE) was the first pioneering method used to explore the genetic population structure and the phylogenetic relationships among T. cruzi natural stocks (Toyé, 1974; Miles et al. 1978) and has been applied to the largest sample of stocks. Valuable information from MLEE studies showed that T. cruzi undergoes predominantly clonal evolution and is distributed into 2 main phylogenetic subdivisions (Tibayrenc, 1995; Souto et al. 1996). Other markers confirmed this subdivision of the taxon into 2 major lineages (Souto et al. 1996; Brisse, Barnabé & Tibayrenc, 2000; Henriksson et al. 2002) which were subsequently referred to as T. cruzi I and II (Momen, 1999). It should be emphasized that this subdivision into 2 main lineages remains controversial (Machado & Ayala, 2001). Moreover, even if this clustering proves to be reliable, these genetic subdivisions are very heterogeneous, and their epidemiological specificity is weak. Within T. cruzi II, 5 lesser subdivisions have been identified, for which epidemiological specificity appears to be much sharper (Barnabé, Brisse & Tibayrenc, 2000; Brisse et al. 2000).
As MLEE remains a basic and simple technique to assess the phylogenetic position of new T. cruzi stocks, the present work aims to exploit the abundant MLEE information for evaluation of the impact of the richness of such information on the robustness of isoenzyme-based phylogenetic reconstruction.
MATERIALS AND METHODS
Stock samples
Thirty-one stocks, representative of the whole known phylogenetic diversity of T. cruzi were selected according to a neighbour-joining tree derived from 262 different genotypes previously identified by using 21 enzyme loci (Barnabé et al. 2000). Fifteen stocks were attributed to T. cruzi I and presented Jaccard's genetic distances (Jaccard, 1908) ranging from 0·05 to 0·57 (mean=0·25±0·11). The 16 other stocks belonged to the 5 different subdivisions of T. cruzi II previously described and their genetic distances ranged from 0·07 to 0·75 (mean=0·39±0·16). The average Jaccard's distance between the two lineages was 0·75±0·07. This selection includes 7 reference stocks commonly used in many laboratories, of which 3 correspond to the early described zymodemes I, II and III (Miles et al. 1978). Two T. cruzi stocks (A83 and A276 isolated from sylvatic mammals in French Guiana) were introduced as outgroups, because they fell out of all other T. cruzi stocks in phylogenetic reconstruction (Lewicka et al. 1995). Tables 1 and 2 summarize the origin and the isoenzyme genotypes of the stocks respectively.
Genetic analysis
Genetic analysis was developed from genotype matrices built with increasing numbers of randomly selected loci (6, 9, 12, 15, 18 and 21). Two additional analyses were done based on the 9 least and most variable loci, respectively. Lastly, analyses were performed on 5 and 9 loci, for which cluster-specific patterns had been previously identified (synapomorphic characters) by Barnabé et al. (2000). Table 3 gives the list of the different sets of loci analysed.
Polymorphism rate (P) and average number of alleles by locus (A) were calculated for each locus sample, based on the hypothesis that T. cruzi is diploid (Lanar, Levy & Manning, 1981; Tibayrenc, Cariou & Solignac, 1981). Observed heterozygosity (Ho) and Nei's mean genetic diversity were calculated for each sample. Clonal diversity (probability that 2 stocks have different genotypes in the population under survey) was measured by Whittam's index (Whittam, 1989): d=n(1−[sum ]Xi2)/(n−1), with Xi=frequency of each multilocus genotype and n=number of individuals.
Phenetic and cladistic analysis were processed for each set of loci. Unweighted Pair-Group Method with Arithmetic Averages (UPGMA) method (Sneath & Sokal, 1973) using Jaccard's distances (Jaccard, 1908) and Wagner analysis (Felsenstein, 1985) were used to cluster the stocks from the different distance matrices. Robustness of the nodes was statistically tested by bootstrap analysis (Felsenstein, 1985) using the PHYLIP package (Felsenstein, 1993).
RESULTS
Isoenzyme variability detected with different numbers of loci
Table 4 shows the indices of genetic variability observed in the different samples. As previously recorded (Barnabé et al. 2000), Mdh was the only monomorphic locus and consequently polymorphism rates were close to 1·00 for each sample. The average number of alleles was logically lower for the set of the 9 least variable loci when compared with the 9 most variable loci (Kruskal–Wallis test, P=3×10−4). In all cases the observed heterozygosity was considerably lower than the expected heterozygosity. This deficit of heterozygous genotypes is a classical indication of clonal population structure (Tibayrenc et al. 1986). Heterozygous genotypes are very scarce in T. cruzi I, whereas T. cruzi II shows several examples of fixed heterozygosity at the Gpi, Idh, Pgm and 6pgdh loci, another classical manifestation of clonal population structure. The number of different genotypes increased when higher numbers of randomly selected loci were used, although most genotypes detected using 21 loci were also identified with lower numbers of loci. When 5 synapomorphic-pattern loci discriminating T. cruzi I and T. cruzi II were applied, only 10 different genotypes were individualized. The clonal diversity was not very different between the sample sets analysed: the probability that 2 stocks have different genotypes was >0·96 in all cases except for the analysis with the 5 synapomorphic-pattern loci for which this index was lower (0·84).
Comparison of clustering analyses produced by different numbers of loci
In agreement with previous results (Tibayrenc, 1995; Souto et al. 1996; Barnabé et al. 2000), the UPGMA and the cladistic analysis of the present sample of stocks using 21 loci identified 2 main subdivisions T. cruzi I and T. cruzi II, among all the studied stocks (bootstrap values: T. cruzi I=0·88, T. cruzi II=0·93). For lower numbers of randomly selected loci, the two groups were generally well identified by the UPGMA analysis, and the stocks A83 and A276 were always clustered apart. Few exceptions were seen by analysing 6 and 9 randomly selected loci, in which CAN III zymodeme 3 was outside T. cruzi II. Moreover, when only 6 loci were used, 3 other stocks were wrongly clustered. Cladistic analysis was performed using the A83 stock as outgroup (Fig. 1). As for the UPGMA analysis, the same stocks were clustered in T. cruzi I and T. cruzi II respectively, except when only 6 randomly selected loci were used. Bootstrap values [ges ]0·73 were obtained for both T. cruzi I and II when either 15, 18 or 21 randomly selected loci were used (Table 5).
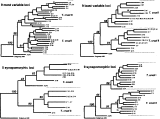
Fig. 1. Phylogenetic Wagner networks obtained by PHYLIP package from matrices of presence/absence of isoenzyme characters using different numbers of loci selected at random. Thirty-one stocks, representative of the genetic variability of Trypanosoma cruzi, were analysed. The stock A83 was selected as outgroup. Bootstrap values of the upper branches are indicated in bold. At the lower level of the tree only bootstrap values [ges ]0·50 are indicated. Numbers of different isoenzyme genotypes correspond to those listed in Table 1.
We compared the results obtained for 4 sets of 9 loci selected according to the following different criteria (i) randomly selected; (ii) the 9 most variable loci; (iii) the 9 least variable loci, and (iv) 9 synapomorphic-pattern loci (Figs 1 and 2). The general topology of UPGMA trees was identical to its corresponding cladistic tree in all cases. Bootstrap values for T. cruzi I and II were very low with (i) since these values were higher with (ii) and even more with (iii). With (iv), these values became comparable to those obtained with 21 randomly selected loci (Table 5). With 5 synapomorphic-pattern loci (Fig. 2) T. cruzi I and II were clearly identified (bootstrap values=0·63 and 0·90 respectively) but they were not distinguished with 6 randomly selected loci (Fig. 1).
Fig. 2. Phylogenetic Wagner networks obtained by PHYLIP package from matrices of presence/absence of isoenzyme characters using different set of loci: the 9 most and least variable loci of 21 studied loci, 5 and 9 loci bearing synapomorphic allozyme characters previously identified by Barnabé et al. (2000). Thirty-one stocks representative of the genetic variability of Trypanosoma cruzi were analysed. The stock A83 was selected as outgroup. Bootstrap values of the upper branches are indicated in bold. At the lower level of the tree only bootstrap values [ges ]0·50 are indicated. Numbers of different isoenzyme genotypes correspond to those listed in Table 1.
Average values of bootstrap were compared (Table 5). They increased according to the number of loci among both T. cruzi I and II, and with all sets of loci, except when 5 synapomorphic-pattern loci were used, when they were lower among T. cruzi I than among T. cruzi II. These results were confirmed by the observation of a higher percentage of bootstrap values [ges ]0·50 within T. cruzi II than within T. cruzi I.
DISCUSSION
The present analysis corroborates the hypothesis that T. cruzi is subdivided into 2 main phylogenetic lineages, T. cruzi I and II (Tibayrenc, 1995; Souto et al. 1996; Momen, 1999). This hypothesis has been challenged by sequence analyses based on limited numbers of genes (Machado et al. 2001). At this step, it can be said that multilocus markers such as isoenzymes, RAPD or microsatellites (Oliveira et al. 1998) all give convergent results. Possibly the sequencing of a higher number of genes will make it possible to settle this debate. However, the goal of the present work was more specifically to evaluate the impact of the genetic information available on this picture, and on the number of multilocus genotypes observed. Multilocus analysis is costly and time-consuming and, in some studies, it could be enough to use a limited number of loci. The different sets of loci were applied to 31 stocks belonging to the 2 T. cruzi lineages, selected to be representative of the whole genetic diversity within each lineage. Even with only 6 randomly selected loci, high number of different genotypes was identified (78% of the number of genotypes identified using 21 loci). In order to test whether this result was obtained by chance, 3 other sets of 6 loci selected at random were examined. The number of different multilocus genotypes was constantly high, ranging from 20 to 27 out of 31 stocks (data not shown). These results show that a limited set of loci is sufficient to uncover much of the total genetic diversity of T. cruzi. However, this result does not hold when phylogenetic analysis is considered. Even if most analyses based on limited sets of loci show the subdivision into T. cruzi I and II, robustness of the nodes evaluated by bootstrap analysis is not the same. An informative, although predictable result was obtained by the use of 9 loci previously identified as bearing synapomorphic characters for the principal and lesser subdivisions recorded within T. cruzi (Barnabé et al. 2000). This limited set of loci can be proposed as bringing the richest information for the lowest amount of work, since it is able to both uncover an important part of the genotype diversity of the sample and also to design a robust phylogenetic picture. Moreover, this set of loci can be recommended to type T. cruzi stocks, which is crucial for both epidemiological surveillance and for any experimental study.
The fact that even limited sets of loci show almost constantly the presence of the two main phylogenetic lineages within T. cruzi is a consequence of the strong linkage disequilibrium between T. cruzi I and T. cruzi II constantly observed in T. cruzi natural populations, and is consistent with the hypothesis that this parasite undergoes predominantly clonal evolution (Tibayrenc et al. 1986) despite occasional events of horizontal gene transfer (Bogliolo, Lauria-Pires & Gibson, 1996; Carrasco et al. 1996; Barnabé et al. 2000; Machado et al. 2001). As previously noted (Brisse et al. 1998, 2000), no clear lesser subdivisions are seen within T. cruzi I, whereas additional structuring within T. cruzi II is suggested by higher bootstrap values in this last group.
The picture of subdivision into 2 main lineages, of which 1 (T. cruzi II) is further subdivided into 5 lesser subdivisions, and the hypothesis that this pattern is the result of predominantly clonal evolution becomes clearer as richer genetic information (increased number of isoenzyme loci) is used in the analysis. The congruence principle (Avise et al. 1994) suggests that this picture and the underlying hypothesis are robust.
The presence of some horizontal gene transfer, or hybridization events, tends to cloud the discreteness of T. cruzi genetic subdivisions. From a purist cladistic view, they may not deserve the name of ‘true’ clades, and the isoenzyme markers that characterize them may not be considered as ‘true’ synapomorphic characters. However, they do correspond to the concept of ‘discrete typing units’ or DTUs (Tibayrenc, 1999), sets of multilocus genotypes that are genetically more related to each other than to any other multilocus genotype, and that are identifiable by one or more specific genetic markers or ‘tags’. The DTUs represent relevant and robust units of analysis for all epidemiological and applied studies dealing with pathogenic microorganisms.
Our study makes it possible to illustrate a general point, frequently neglected in many phylogenetic studies. Bootstrap results are always given under the form of rough figures with an artificial border between significant and non-significant. Now bootstrap is only a statistical test, for which power is highly dependent upon the richness of information available (Tibayrenc, 1999). In the present work, for the same biological sample and the same genetic typing method, robustness of the results was greatly influenced by the number of loci. In many studies, low bootstrap values could well not indicate lack of phylogenetic subdivisions, but could imply a statistical type II error (lack of power of the test due to insufficient data).