



Introduction
Dientamoeba fragilis is a trichomonad parasite that resides in the human bowel (Stark et al., Reference Stark, Barratt, Chan and Ellis2016). Despite being found on every inhabited continent and being the most prevalent trichomonad parasite to infect humans (Stark et al., Reference Stark, Barratt, Chan and Ellis2016), relatively little genetic research has been published on this unusual organism. The role of D. fragilis in human gut health is under debate, and its pathogenic potential is under investigation (Barratt et al., Reference Barratt, Harkness, Marriott, Ellis and Stark2011; Jokelainen et al., Reference Jokelainen, Hebbelstrup Jensen, Andreassen, Petersen, Roser, Krogfelt, Nielsen and Stensvold2017). Unfortunately, current culture systems for D. fragilis are xenic, usually containing a poorly defined mixture of bacteria, which greatly hampers research on this organism. Several xenic culture systems exist for D. fragilis, the most commonly used is a biphasic Loefflers serum slope overlaid with PBS and supplemented with rice starch granules (Barratt et al., Reference Barratt, Banik, Harkness, Marriott, Ellis and Stark2010; Munasinghe et al., Reference Munasinghe, Stark and Ellis2012). Long term cryopreservation of D. fragilis trophozoites has been reported using various concentrations of dimethyl sulfoxide (DMSO) and glucose, though these methodologies have been used with varying success (Dwyer and Honigber, Reference Dwyer and Honigber1971; Sawangjaroen et al., Reference Sawangjaroen, Luke and Prociv1993; Barratt et al., Reference Barratt, Banik, Harkness, Marriott, Ellis and Stark2010).
D. fragilis has co-evolved with the natural flora of the bowel, which it voraciously grazes upon by phagocytosis. These prokaryotes undoubtedly satisfy an important nutritional requirement for growth of this protozoan in vivo and in vitro. Indeed, pioneering studies by Brug (Reference Brug1936) and Jacobs (Reference Jacobs1953) confirmed a strong dependence on live bacteria as a food source for cultured D. fragilis trophozoites, specifically noting that dead bacteria were insufficient for sustained trophozoite growth.
The bacteria in D. fragilis cultures complicate research on this parasite, including animal studies aiming to elucidate its pathogenicity (Munasinghe et al., Reference Munasinghe, Vella, Ellis, Windsor and Stark2013), as the presence of these bacteria is difficult to control for. Additionally, the presence of bacterial DNA in DNA extracts from D. fragilis cultures represents a significant challenge for whole genome sequencing. Bacterial cells which generally outnumber D. fragilis trophozoites in culture, are present in the cytoplasm of the protozoan due to its grazing and are difficult to remove by methods of washing and centrifugation. Consequently, bacteria-derived genomic DNA is difficult to exclude from D. fragilis culture extracts and competes with that of D. fragilis in sequencing experiments. These difficulties are increased by the fact that the D. fragilis genome, like that of other trichomonads, is probably extremely large for a protozoan and highly repetitive (Barratt et al., Reference Barratt, Gough, Stark and Ellis2016). The net result is a fragmented assembly consisting of partial bacterial genomes, some contigs derived from D. fragilis, and many contigs for which the origin cannot be accurately determined. In addition, many early branching protozoa, including the trichomonads, are known recipients of bacterial genes obtained via lateral gene transfer (Barratt et al., Reference Barratt, Gough, Stark and Ellis2016). This also complicates sequencing experiments involving D. fragilis, and was an issue for the D. fragilis transcriptome sequencing project where a proportion of the transcripts could not be confidently assigned to a prokaryotic or eukaryotic origin (Barratt et al., Reference Barratt, Cao, Stark and Ellis2015). Reducing the complexity of bacteria in D. fragilis cultures would greatly assist these downstream sequencing experiments.
Preliminary studies defining the microbial flora in D. fragilis cultures were conducted by Barratt et al. (Reference Barratt, Banik, Harkness, Marriott, Ellis and Stark2010) using standard phenotypic microbiological techniques. Several bacterial species were identified, with Escherichia coli predominating in each isolate (Barratt et al., Reference Barratt, Banik, Harkness, Marriott, Ellis and Stark2010). While this is not surprising, these classical microbiological methods do not capture the complete diversity of a sample, particularly in complex samples such as those derived from the human gut. Moreover, metagenomic analyses have revealed that at least 2100 bacterial species inhabit the gastrointestinal tract of humans, the vast majority of which cannot be cultured using traditional laboratory techniques (Eckburg et al., Reference Eckburg, Bik, Bernstein, Purdom, Dethlefsen, Sargent, Gill, Nelson and Relman2005; Hugon et al., Reference Hugon, Dufour, Colson, Fournier, Sallah and Raoult2015). The sequencing of metagenomes has become possible due to rapid advancements in DNA sequencing and bioinformatics enabling large scale projects that provide unprecedented insight into the diversity of complex microbial communities (Hugenholtz, Reference Hugenholtz2002; Tringe et al., Reference Tringe, von Mering, Kobayashi, Salamov, Chen, Chang, Podar, Short, Mathur, Detter, Bork, Hugenholtz and Rubin2005).
The primary aim of this present study was to investigate ways that would reduce the number and complexity of bacterial species present in D. fragilis cultures that in turn would facilitate the generation of genome sequencing data from D. fragilis using a metagenomics approach. Initially, the bacterial diversity of clinical as well as previous D. fragilis in vitro culture samples was investigated in order to define those bacterial species associated with D. fragilis growth. Specific bacteria were then systematically eliminated from D. fragilis cultures using antibiotic treatments and the impact of these treatments on the microbial community was assessed by 16S ribosomal DNA bacterial diversity profiling. Following the reduction of bacterial species in culture, whole genome sequencing was conducted. A metagenomic approach to genome assembly was applied, resulting in the identification of the previously uncharacterized D. fragilis 28S ribosomal subunit gene. This sequencing represents the first step and a proof of concept towards the study of the entire D. fragilis genome.
Methodology
Collection of stool specimens and maintenance of D. fragilis cultures
Isolates A, E, and V were isolated and cultured during a previous study and stored at −80 °C (Barratt et al., Reference Barratt, Banik, Harkness, Marriott, Ellis and Stark2010). Isolate Q was cultured during this study from the stool of a symptomatic patient who presented at St. Vincent's Hospital, Sydney for investigation of bowel complaints. This isolate was maintained routinely in the diagnostic laboratory (SYDPath) at St Vincent's Hospital, Sydney, using the culture techniques described below. Faecal samples were obtained from symptomatic patients carrying D. fragilis and the presence or absence of bacterial and protozoan pathogens was confirmed using the Genetic Signatures EasyScreen enteric parasite detection kit and the Genetic Signatures EasyScreen enteric prokaryote detection kit (Stark et al., Reference Stark, Roberts, Ellis, Marriott and Harkness2014). A second unpreserved stool sample was obtained from the D. fragilis positive patient and ~10 mg of stool was placed onto a 5 mL inspissated serum slope (prepared as described below) overlaid with 3 mL of PBS containing 0.1 g of refined starch granules. Care was taken to minimize exposure of D. fragilis faecal samples to oxygen in order to maximize the success of the cultures.
To prepare the inspissated serum slopes, 5 mL volumes of Loeffler's slope serum were aliquoted into McCartney bottles under sterile conditions. The bottles were placed at a 30–45° angle during insipissation. The slopes were then autoclaved using a dry sterile cycle of 89 °C for 15–20 min and allowed to cool and solidify at room temperature, before storage at 4 °C.
Following inoculation with stool the cultures were placed in an anaerobic jar under microaerophilic conditions (0.2% O2, 9.9% CO2, 5% H2 and 84.9% Ns) using the Anoxomat apparatus (Advanced Instruments). Cultures were incubated at 37 °C for 48 h, and then examined for D. fragilis trophozoites using phase contrast microscopy at 400× magnification. This was performed routinely on positive cultures to follow their longevity. Approximately 250 fields of view were inspected for each slide, with positive identification of D. fragilis growth based upon the characteristic morphology of D. fragilis. D. fragilis cultures were passaged into fresh media and incubated at 37 °C under microaerophilic conditions on a 48–72 h weekly timetable.
DNA extraction from faecal and culture samples
DNA was extracted from 10 faecal samples confirmed positive for D. fragilis trophozoites using multiplex qPCR (Genetic Signatures), and 10 control faecal samples, all of which were considered free of gastrointestinal pathogens via multiplex qPCR (Genetic Signatures). The extraction of DNA from D. fragilis cultures and stool was performed using a Qiagen DNA extraction robot (EZ1). Briefly, 250 µl G2 buffer, 10 µl Proteinase K and 10 mg of stool or culture sample were added to an Eppendorf tube and vortexed for 30 s to mix thoroughly. Samples were then placed on a heating block at 95 °C for 10 min. Samples were then spun at 13 000 rpm for 2 min. Next, 200 µL of supernatant for each sample was removed and used for DNA extraction using the EZ1 and the Qiagen EZ1 DNA tissue kit, following the manufacturer's extraction protocol.
Bacterial diversity profiling
Genomic extracts of DNA (20 µL) derived from the two faecal sample groups (10 stool containing D. fragilis and 10 negative stool controls) and four cultured isolates were submitted to the Australian Genome Research Facility (AGRF) service provider for amplicon sequencing of the V3–V4 341F – 806R region of the 16S rDNA gene using propriety PCR primers and the Illumina MiSeq platform. The DNA samples were prepared according to the service providers instructions, at a concentration of greater than 10 ng μl−1 and with A260/A280 ratios between the range of 1.6–1.9. Upon receipt of these DNA samples, AGRF performed a polymerase chain reaction (PCR) targeting the V3–V4 341F – 806R region of the 16S ribosomal DNA using the forward primer sequence 5′-CCTAYGGGRBGCASCAG-3′ and the reverse primer sequence 5′-GGACTACNNGGGTATCTAAT-3′. Forward and reverse primer overhangs were also appended to the primer pair sequences for compatibility with Illumina index and sequencing adapters (forward: 5′-TCGTCGGCAGCGTCAGATGTGTATA AGAGACAG-3. Reverse: 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′). The amplicons from each sample were then sequenced in multiplex, on the Illumina MiSeq platform, utilizing Illumina's Nextera XT v2 indices and paired end sequencing chemistry. In addition to the paired-end .fastq sequence files generated, a report was generated including a description of the operational taxonomic units (OTUs), their relative abundance and raw read counts for all prokaryotic taxa detected in each sample. One way Analysis of Similarity (ANOSIM) based on Bray–Curtis distance of this microbiome data was performed using PAST V3.15 (Hammer et al., Reference Hammer, Harper and Ryan2001).
Antibiotic treatment of xenic D. fragilis cultures
Antibiotic treatment regimens were used to systematically remove bacteria based on the information received from the diversity profile. Antibiotics were selected depending on their Gram identification and aerobic requirements. Meropenem (Ranbaxy) was added to D. fragilis cultures at a concentration of 10 µg mL−1 which exceeds the sensitive/intermediate (S/I) breakpoint of ⩽2 mg L−1 recommended by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (Giske et al., Reference Giske, Martinez-Martinez, Canton, Stefani, Skov, Glupczynski, Nordmann, Wootton, Miriagou, Simonsen, Zemlickova, Cohen-Stuart and Gniadkowski2017). Vancomycin (Ranbaxy) was added to a concentration of 10 µg mL−1, exceeding the EUCAST MIC break point for Vancomycin of >2 mg L−1 (Giske et al., Reference Giske, Martinez-Martinez, Canton, Stefani, Skov, Glupczynski, Nordmann, Wootton, Miriagou, Simonsen, Zemlickova, Cohen-Stuart and Gniadkowski2017). Preliminary experiments showed that removal of Gram negative bacteria from the D. fragilis cultures was lethal to D. fragilis and these observations led to the incorporation of a drug-resistant Klebsiella pneumonia into the cultures in the following way.
Immediately following the addition of an antibiotic to culture, two colonies of KPC-2 carbapenem resistant K. pneumonia originally sourced from Villegas et al. (Reference Villegas, Lolans, Correa, Suarez, Lopez, Vallejo and Quinn2006), were picked from a Blood agar plate and added to the cultures. Ten passages with antibiotic treatment were performed before cultures were resubmitted to AGRF to assess the impact of the antibiotic treatment. Antibiotic treated cultures were passaged in triplicate on the same 48–72 hour schedule, with antibiotics and KPC-2 K. pneumoniae added after each passage. D. fragilis trophozoite counts were assessed via qPCR that was validated against a titration of known cell counts (Chan et al., Reference Chan, Barratt, Roberts, Phillips, Šlapeta, Ryan, Marriott, Harkness, Ellis and Stark2016; Gough et al., Reference Gough, Ellis and Stark2019). Throughout the experiment untreated cultures (in triplicate) were also passaged as a negative control to the antibiotic treatment.
Genome sequencing and bioinformatic analysis/validation
Specific details, scripts and parameters regarding the sequencing and bioinformatic methodologies are provided in the Supplementary Materials (Sup. 1). In summary, an Illumina gDNA shotgun library was prepared by AGRF using DNA extracted from an antibiotic treated D. fragilis culture. The library was then sequenced on an Illumina HiSeq 2500 platform producing 125 bp paired end reads. Reads were then trimmed, filtered and assembled using the metagenomic software metaSPAdes (Nurk et al., Reference Nurk, Meleshko, Korobeynikov and Pevzner2017). In order to confirm the presence of D. fragilis reads prior to assembly, trimmed and filtered reads were mapped using Bowtie2 against a previously published D. fragilis transcriptome (Barratt et al., Reference Barratt, Cao, Stark and Ellis2015), as well as the Trichomonas vaginalis shotgun genome (Carlton et al., Reference Carlton, Hirt, Silva, Delcher, Schatz, Zhao, Wortman, Bidwell, Alsmark, Besteiro, Sicheritz-Ponten, Noel, Dacks, Foster, Simillion, Van de Peer, Miranda-Saavedra, Barton, Westrop, Müller, Dessi, Fiori, Ren, Paulsen, Zhang, Bastida-Corcuera, Simoes-Barbosa, Brown, Hayes, Mukherjee, Okumura, Schneider, Smith, Vanacova, Villalvazo, Haas, Pertea, Feldblyum, Utterback, Shu, Osoegawa, de Jong, Hrdy, Horvathova, Zubacova, Dolezal, Malik, Logsdon, Henze, Gupta, Wang, Dunne, Upcroft, Upcroft, White, Salzberg, Tang, Chiu, Lee, Embley, Coombs, Mottram, Tachezy, Fraser-Liggett and Johnson2007).
To identify contigs within the assembly, the metaSPAdes scaffold output was visualized using Bandage (Holt et al., Reference Holt, Schultz, Wick and Zobel2015). Contigs containing rDNA sequence were identified by a BLASTN search within Bandage using the T. vaginalis 28S rDNA (NW_001581582). Primers (forward: 5′-GGAGCTTGTGGCTTAATTTGA-3′. reverse: 5′-GTCGGCATAGTTTAAGGTAGG-3′) were designed to amplify a section of the rDNA repeat by a long-range PCR using the following reagents: 10 µl of Ranger polymerase buffer (Bioline), 2 µl of 10 m forward primer, 2 µl of 10 m reverse primer, 2.5 µl of DMSO, 1 µl of Ranger polymerase (Bioline), 20.5 µl H20 and 2 µl of D. fragilis gDNA template. The reaction was then subjected to the following cycling conditions: 95 °C for 3 min, followed by 30 cycles of 98 °C for 10 s and 58 °C for 8 min. The PCR product was then sequenced using the Nextera FLEX sample preparation and MiSeq Nano V2 next generation sequencer (Illumina). The reads were trimmed and filtered, then assembled using metaSPAdes.
Results
D. fragilis trophozoite growth
D. fragilis trophozoites were identified according to their morphology, namely a trophozoite diameter of 5–15 µm, ranging from spherical to amoeboid in shape, often accompanied by the presence of ingested starch granules which produce a refractile appearance under light microscopy (Fig. 1). Cell counts reached a peak of 250 trophozoites per μl of culture, although numbers fluctuate depending on the time cultures are observed and their position in the cell life cycle.

Fig. 1. Phase contrast microscopy of cultured D. fragilis trophozoites (isolate Q). (A) Displays a D. fragilis trophozoite of typical morphology with visible digestive vacuoles. (B) Shows a D. fragilis trophozoite of amoebic morphology. (C) Displays a large D. fragilis trophozoite with ingested starch granules. An example of trophozoite size variation can be seen in D which contains two trophozoites with ingested starched granules.
Bacterial diversity of faecal samples
Over 208 individual OTUs (Observational Taxonomic units) were identified in the ten control and ten D. fragilis positive faecal samples collected from St Vincent's Hospital. Despite this large number, total bacterial abundance was dominated by 20 OTUs, with the remaining 188 having a total abundance of less than 1% across the 20 samples in total. Of the 20 most abundant OTUs, 17 were identified as Gram-negative which is a common observation reported in other studies of the human gut microbiome (Arumugam et al., Reference Arumugam, Raes, Pelletier, Le Paslier, Yamada, Mende, Fernandes, Tap, Bruls, Batto, Bertalan, Borruel, Casellas, Fernandez, Gautier, Hansen, Hattori, Hayashi, Kleerebezem, Kurokawa, Leclerc, Levenez, Manichanh, Nielsen, Nielsen, Pons, Poulain, Qin, Sicheritz-Ponten, Tims, Torrents, Ugarte, Zoetendal, Wang, Guarner, Pedersen, de Vos, Brunak, Doré, Meta, Antolín, Artiguenave, Blottiere, Almeida, Brechot, Cara, Chervaux, Cultrone, Delorme, Denariaz, Dervyn, Foerstner, Friss, van de Guchte, Guedon, Haimet, Huber, van Hylckama-Vlieg, Jamet, Juste, Kaci, Knol, Kristiansen, Lakhdari, Layec, Le Roux, Maguin, Mérieux, Melo Minardi, M'Rini, Muller, Oozeer, Parkhill, Renault, Rescigno, Sanchez, Sunagawa, Torrejon, Turner, Vandemeulebrouck, Varela, Winogradsky, Zeller, Weissenbach, Ehrlich and Bork2011). PAST statistical software was used to analyse the significance of OTU presence between the two sample groups. One way ANOSIM showed a significant difference in bacterial diversity in D. fragilis positive samples compared to the control group (P = 0.0203) although separation between the groups was low (R = 0.1353) (Fig. 2). Enterobacteriaceae were four times more abundant in the control group than D. fragilis positive samples. Conversely, Akkermansia muciniphila was five times more abundant in D. fragilis positive samples. Prevotella copri was also seen in incredibly high abundance in two samples from the D. fragilis positive group (51% and 19%), an abundance greater than that seen in the two control group samples in which it was found (3.3% and 1.3%). Relative abundances of the 20 most prominent OTUs can be seen in Table 1.
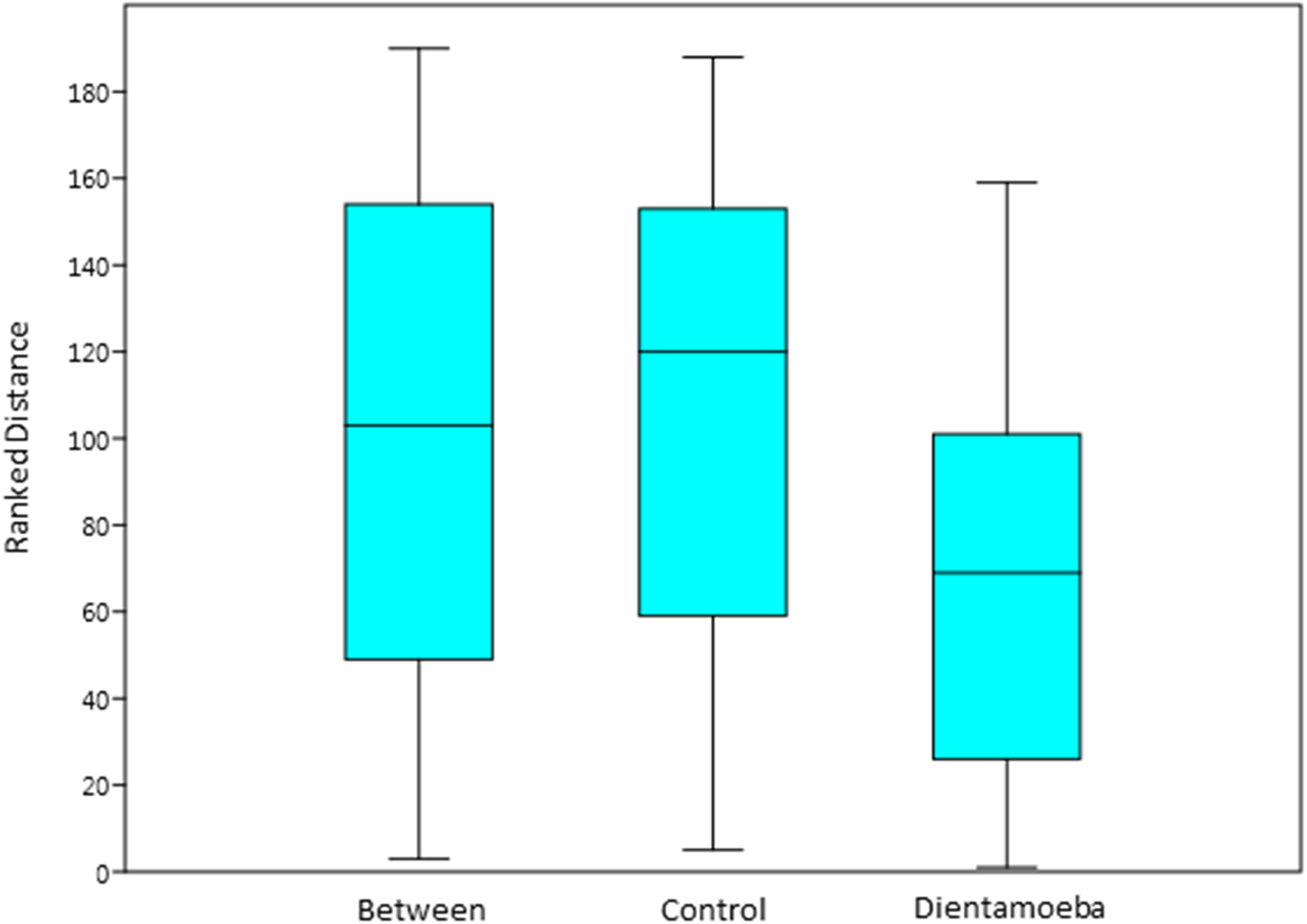
Fig. 2. One way analysis of similarity (ANOSIM) based on Bray–Curtis distance for bacterial diversity across control faecal samples and D. fragilis positive samples. ANOSIM showed a significant difference in bacterial diversity in D. fragilis positive samples compared to the control group (P = 0.0203) although separation between the groups was low (R = 0.1353).
Table 1. Twenty most abundant OTUs for control and D. fragilis positive faecal samples. OTUs are defined to their most specific level provided by AGRF 16S sequencing. O: order, F: family, G: genus, S: species. C1–10 = control samples 1–10, D1–10 = D. fragilis positive samples 1–10

Bacterial diversity of long term D. fragilis cultures
Isolate Q cultures exhibited constant, rigorous growth for the duration of the experiment and provided sufficient material for DNA extraction. Individual D. fragilis cells exhibited mostly typical morphology (pleomorphic, ranging in size from 10 to 15 µm), with the majority of cells displaying evidence of recent ingestion of starch granules, examples of which can be seen in Fig. 1.
A diverse range of bacterial species were identified across isolates A, E, Q and V, although bacterial diversity was significantly dominated by members of the Enterobacteriaceae family, and the Prevotella and Bacteroides genera. These three made up 65% of all bacterial reads across the four isolates. OTUs equal to or greater than 1% of total abundance can be seen in Fig. 3.

Fig. 3. Bacterial diversity in long term D. fragilis cultures. Bacterial reads are classified to their most specific definition according to 16S ribosomal sequencing. Legend: O, order, F, family, G, genus, S, species. OTUs of <1% have been omitted but can be found in the supplementary information. Culture names are provided above each pie chart.
Meropenem treatment
Introduction of meropenem to cultures reduced anaerobic Gram-negative growth but also led to protozoan death after 5 days. Inclusion of the meropenem resistant KPC-2 K. pneumoniae into cultures before meropenem treatment prevented the death of D. fragilis cells beyond 5 days, suggesting some nutritional requirement is provided by Gram negative bacterial cells in culture. 16S bacterial diversity profiling of meropenem treated culture spiked with KPC-2 K. pneumoniae revealed a total of 27 bacterial OTUs present within the culture, a reduction of 12 compared to the 39 total species found in the untreated isolate Q culture. More importantly, 18 of the 27 OTUs identified in the meropenem treated culture had a total relative abundance of less than 1%. The meropenem resistant KPC-2 K. pneumoniae dominated total abundance with 49.1%. Of the nine most abundant OTUs identified in meropenem treated cultures, four were Gram-positive bacteria. Specific abundances for OTUs over 1% can be found in Fig. 4. D. fragilis trophozoite growth was assessed at this point using qPCR. Although trophozoite numbers fell to 2500 trophozoites per mL after meropenem treatment from 7000 trophozoites per mL in the untreated control, trophozoite growth was constant and cultures remained stable for the duration of the experiment.

Fig. 4. Effect of antibiotic treatment on bacterial diversity in D. fragilis cultures. (A) Bacterial diversity post meropenem treatment and (B) bacterial diversity post meropenem and vancomycin treatment. Meropenem reduced bacterial diversity from 18 OTUs over 1% abundance to nine bacterial OTUs. Vancomycin further reduced bacterial diversity to only five OTUs with abundance over 1%.
Vancomycin treatment
Addition of vancomycin to cultures after meropenem treatment eliminated all Gram-positive bacteria from the culture further reducing the total number of bacterial OTUs to 13. Only five OTUs were identified as having a relative abundance greater than 1%. The vast majority of this relative abundance was again dominated by the KPC-2 K. pneumoniae, reaching 78.4% (Fig. 4). Following vancomycin treatment, qPCR revealed D. fragilis trophozoite numbers decreased to an estimated 150 trophozoites per mL by qPCR. Despite the reduced cell count, D. fragilis cultures remained viable and stable for a period of 6 weeks. DNA was extracted from this culture for genomic DNA sequencing.
Structure and general features of the D. fragilis 28S-18S rDNA repeat
Following read filtering and trimming, 48 067 259 paired end reads remained. Mapping to the D. fragilis transcriptome and T. vaginalis shotgun genome revealed 809 813 of those reads mapping to the indexed sequences. This equates to 1.6% of the total reads, with the remaining 98.4% of reads being of prokaryotic origin. Assembly of the filtered and trimmed reads generated 32 427 contigs, the longest being 1 235 710 bp in length, identified via BLASTN as the Lachnoclostridium sp. YL32 complete genome (GenBank accession number: CP 015399.2). Following metagenome assembly, an 8 Kb contig was identified in Bandage using a BLASTN search with a T. vaginalis 28S rDNA sequence.
In order to confirm the sequence of the rDNA contig, long range PCR amplified a product of ~8 Kb, although following sequencing, a contig of only 4702 bp (Sup. 2.) could be assembled, the structure of which can be found in (Fig. 5). The contig contained 1501 bp of 18S rDNA, 85 bp of ITS1, 161 bp of 5.8S rDNA, 63 bp of ITS2, 2847 bp 28S rDNA and finally a 45 bp IGS partial sequence (GenBank accession number: MK526886). BLASTN analysis showed the D. fragilis 18S, ITS1 and ITS2 sequences to be highly homologous with those previously deposited in Genbank (JQ677148, DQ233461). The D. fragilis 28S rDNA returned similarities of 85% and 79% to the Histomonas meleagridis (HM185490) and T. vaginalis 28S (NW_001581582) rDNA sequences respectively.

Fig. 5. D. fragilis ribosomal DNA repeat gene structure. D. fragilis rDNA gene structure and arrangement following sequencing of long range PCR amplicon. The three ribosomal RNA genes (18S, 28S and 5.8S), internal transcribed spacers (ITS1 and 2) and (IGS) are shown.
Discussion
Transcriptome studies on D. fragilis trophozoites suggest that this species expresses proteins associated with virulence (Barratt et al., Reference Barratt, Cao, Stark and Ellis2015). However increasing recognition of the complexity of the host/parasite/microbiome relationship indicates commensal bacteria and probiotics play some role in the outcome of protozoan infections (Bär et al., Reference Bär, Phukan, Pinheiro and Simoes-Barbosa2015; Partida-Rodriguez et al., Reference Partida-Rodriguez, Serrano-Vazquez, Nieves-Ramirez, Moran, Rojas, Portillo, Gonzalez, Hernandez, Finlay and Ximenez2017; Leung et al., Reference Leung, Graham and Knowles2018). It is currently impossible to isolate D. fragilis from faecal samples without co-culturing of the host's gastrointestinal microbiota. However, there has been no investigation examining the associations between D. fragilis infection and host gastrointestinal microbiota. While this association was not the primary aim of this study due to the limited sample size, it was necessary to assess bacterial diversity of in vivo and in vitro samples in order to select an appropriate bacterial species to spike cultures with in order to prevent trophozoite death.
Host cells within the human gastrointestinal tract are outnumbered by bacterial cells to a factor of 10 (Bull and Plummer, Reference Bull and Plummer2014). The majority of these bacterial cells are considered strict anaerobes, outnumbering aerobic bacteria by 100 fold (Harris et al., Reference Harris, Reddy and Carter1976). Although a wide range of bacteria have been identified in the human gut to date, the typical host gut microbiome is dominated by two phyla, Firmicutes and Bacteroidetes, the vast majority of which are commensal or beneficial to the host (Lozupone et al., Reference Lozupone, Stombaugh, Gordon, Jansson and Knight2012). This allows the gut to attempt to maintain homeostasis via functional redundancy, where multiple microbial groups carry out the same functional process under the same conditions, allowing for bacterial diversity between individual hosts without compromising complete function for a particular host (Sekirov et al., Reference Sekirov, Russell, Antunes and Finlay2010; Lozupone et al., Reference Lozupone, Stombaugh, Gordon, Jansson and Knight2012). This delicate network that is formed from the amalgamation of host/bacterial interactions is vital to human health and disruption to this complex balancing act has been associated with a myriad of human diseases including inflammatory bowel disease (IBD) (Dicksved et al., Reference Dicksved, Halfvarson, Rosenquist, Jarnerot, Tysk, Apajalahti, Engstrand and Jansson2008), obesity (Ley et al., Reference Ley, Turnbaugh, Klein and Gordon2006), and cancer (Roy and Trinchieri, Reference Roy and Trinchieri2017). Additional complexity is added to these networks when the presence of commensal and pathogenic protozoa are considered. What develops is a food chain hierarchy of protozoan apex predators and bacterial prey.
The bacterial microbiomes of two sample groups (10 stool containing D. fragilis and 10 negative stool controls) were sequenced using 16S: V3–V4 341F – 806R on the Illumina MiSeq platform, then compared against each other to understand what bacterial diversity D. fragilis encounters in vivo and to examine if normal bacterial diversity is different in the presence of D. fragilis trophozoites. This information was necessary for the progression of this study, as it was suggested that the initial bacterial composition of in vitro D. fragilis cultures is likely to emulate that found in vivo. DNA was extracted from 10 faecal samples that did not contain D. fragilis and 10 faecal samples that were positive for D. fragilis infection via qPCR. Over 200 individual OTUs were identified between the 20 samples. Bacterial abundance was dominated by 20 OTUs, leaving a large varied collection of auxiliary microbiota, again indicative of gastrointestinal functional redundancy.
Significant differences were seen between the two sample groups (P = 0.02) although overall separation was low with an R value of 0.1353. The sample size used makes association of specific OTUs to D. fragilis infection problematic so we restrict our comments to significant observations.
Major differences between the two sample groups were confined to a limited number of OTUs, primarily Enterobacteriaceae, A. muciniphila and P. copri. The high abundance of A. muciniphila in D. fragilis positive groups is particularly interesting due to A. muciniphila's use as an anti-inflammatory agent (van Passel et al., Reference van Passel, Kant, Zoetendal, Plugge, Derrien, Malfatti, Chain, Woyke, Palva, de Vos and Smidt2011) and has not been observed in similar studies focusing on D. fragilis (O'Brien Andersen et al., Reference O'Brien Andersen, Karim, Roager, Vigsnæs, Krogfelt, Licht and Stensvold2016). Studies have concluded that an increase in A. muciniphila corresponded with a reduction in gastrointestinal inflammation in mice, a finding potentially at odds with the data returned from our study (Caesar et al., Reference Caesar, Tremaroli, Kovatcheva-Datchary, Cani and Bäckhed2015). It is possible that the patients from which the D. fragilis positive faecal samples were collected may have been taking probiotic supplements containing A. muciniphila but this information was not accessible in this study. The overall reduction of Enterobacteriaceae across the D. fragilis positive samples was surprising, considering this OTU was the most abundant in three of the four long term D. fragilis cultures.
Bacterial diversity profiles were established for all D. fragilis cultures available, in order to explore the identity of the bacteria present in long term in-vitro cultures as well as to identify the most effective antibiotic treatments needed to reduce the bacterial diversity present in them. A predominance of the Gram-negative bacteria was seen in these long term cultures where members of the Enterobacteriaceae family, and the Prevotella and Bacteroides genera, contributed 65% of bacterial 16S sequence reads across the four isolates.
On the basis of the 16S bacterial diversity profiles for faecal and long term cultures, meropenem was chosen as the first antibiotic to reduce bacterial diversity within cultures of isolate Q. Meropenem is a broad spectrum antibiotic with activity against a range of Gram-negative and positive bacterial species (Sheikh et al., Reference Sheikh, Pitkin and Nadler1993). This broad spectrum activity was apparent upon first use, as bacterial cell numbers drastically dropped within 4 days and D. fragilis trophozoite numbers followed suit within an additional 2 days, indicating the requirement for a minimum threshold of bacteria needed in D. fragilis culture, an observation supporting earlier studies (Brug, Reference Brug1936; Jacobs, Reference Jacobs1953). It became apparent that without the inclusion of a constant bacterial food source resistant to meropenem activity, trophozoite numbers would never recover.
Meropenem and vancomycin were selected for the first round of treatments due to their broad coverage over Gram-negative and Gram-positive bacteria, both inhibiting bacterial cell wall synthesis with vancomycin also inhibiting RNA synthesis (Watanakunakorn, Reference Watanakunakorn1981; Wiseman et al., Reference Wiseman, Wagstaff, Brogden and Bryson1995). KPC-2 K. pneumoniae was chosen for its specific meropenem resistance and its availability on site. Dixenic culture with K. pneumoniae and Bacteroides vulgatus was previously reported by Chan et al. (Reference Chan, Guan, Mackenzie and Diaz-Mitoma1994). In that study, dixenic Dientamoeba cultures were obtained from the American Type Culture Collection (ATCC), although bacterial composition was only assessed via conventional methods using culture media and standard biochemical tests. Assessing taxonomy via the inoculation of bacteriological media does not offer the same resolution as next generation sequencing, as many bacterial species are not culturable or unable to be distinguished to a taxonomic level through morphology and growth conditions alone. Indeed, Chan et al. (Reference Chan, Guan, Mackenzie and Diaz-Mitoma1994) reported two distinct colony morphologies for B. vulgatus suggesting a higher bacterial diversity than reported. In the study reported here, addition of KPC-2 to the D. fragilis culture had an immediate effect and prevented loss of D. fragilis trophozoites along with a clear reduction in bacterial diversity from the antibiotic treatment.
The elimination of Gram-positive bacteria was the logical next step and vancomycin was selected for its Gram-positive specificity. Not only were all Gram-positive bacterial OTUs eliminated from the culture but overall diversity dropped to an unprecedented level. It must be noted that this result is only indicative of isolate Q, as this was the only in vitro culture available at the time. Any conclusions on D. fragilis’ ability to survive in the absence of Gram negative bacterial cells can only be extrapolated for other isolates at this time.
The incredible coverage that 16S next generation sequencing provides has highlighted the shortcomings of previous investigations into the bacterial diversity found in D. fragilis cultures. Historially, both mono and dixenic D. fragilis cultures have been reported (Balamuth, Reference Balamuth1946; Jacobs, Reference Jacobs1953), although neither of these studies were able to be reproduced and the limitations of the time are apparent once new sequencing technology is considered. What may appear to be one or two bacterial species through traditional techniques is often revealed to be much more complex and diverse through next generation sequencing. Identification of the co-cultured bacterial species was often inaccurate as was the case with Balamuth's study, with their original identification of Clostridium perfringens and Aerobacter aerogenes being later revised to K. pneumoniae and C. perfringens, and then again later reclassifying the anaerobic species to Bacteroides (Chan et al., Reference Chan, Guan and Mackenzie1993). The presence of a Gram positive bacteria in these previous studies is at odds with the findings of this paper, although it is apparent that even with the inclusion of antibiotic treatment, previous studies were not able to confidently exclude the presence of unidentifiable and ‘unculturable’ bacteria, of which a menagerie are present within faecal samples. This uncertainty leaves much room for error and revision.
More recently, a preliminary study identified E. coli as the predominant bacterial species within in-vitro cultures of isolate A, E and V (Barratt et al., Reference Barratt, Banik, Harkness, Marriott, Ellis and Stark2010). Susceptibility studies by (Nagata et al., Reference Nagata, Marriott, Harkness, Ellis and Stark2012) identified several members of the Enterobacteriaceae family and other Gram-negative species within isolates A, E and V using a combination of RapID API strips (Biomeriéux) and 16S sequencing, although this study did not quantify their relative levels of abundance. Due to the diversity found within each culture, where many bacterial species lack published culturing methodologies, standard laboratory techniques are limited for detailed investigations of bacterial community composition.
The final aim of the study was to generate new genome sequence data from D. fragilis following elimination of bacteria from cultures. DNA sequencing showed that bacterial DNA was still present at significant levels even after antibiotic treatment of the cultures, and so very few contigs from D. fragilis were identified in the assembled sequence data. The use of Bandage helped identify a 28S-18S rDNA repeat with homology to T. vaginalis and H. meleagridis. This is likely the most abundant sequence in the D. fragilis cell and as such offered a convenient locus to identify as a proof of concept for this metagenomics approach to sequencing a protozoan genome from mixed microbial reads. There are potentially hundreds of rDNA repeats in each D. fragilis genome and so it is not surprising that this sequence was recovered. The rDNA repeat shares the features typical of many protozoa, a 18S – 5.8S – 28S ribosomal DNA region separated by non-transcribed spacers ITS spacers (Srivastava and Schlessinger, Reference Srivastava and Schlessinger1991). The 28S region of the D. fragilis rDNA repeat has not been reported to date and this data represents a novel addition to the D. fragilis rDNA sequence. The D. fragilis 28s region, while sharing a high degree of homology, is significantly larger than that of the GenBank submitted H. meleagridis 28S partial sequence (1928 bp, HM185490). At 2847 bp, the D. fragilis 28S sequence is similar in length to the T. vaginalis complete 28S sequence (NW_001581582), suggesting that the assembled sequence is indeed represented in its entirety. The intergenic spacer (IGS) could be visualized using Bandage, seen as multiple divergent contigs and paths, typical of a highly repeated region that exhibits a wide degree of variability. Indeed, this variability can be attributed to the failure of metaSPAdes to successfully integrate this region into a single contig covering the entirety of the rDNA repeat. Intergenic spacer regions vary in length from ~2 kb in yeast to 21 kb in mammals and are highly variable even between individuals of the same species (Moss and Stefanovsky, Reference Moss and Stefanovsky1995). Considering a long range PCR product of ~8 Kb was amplified, it can be reasoned that the D. fragilis IGS must be at least 3 kb in size, accounting for the 4.7 kb rDNA contig that was assembled.
Ultimately, this study is the first to confirm that Gram-positive bacteria are not a requirement for D. fragilis growth in vitro. In addition, this study is the first to adopt a metagenomic approach to accurately characterize the microbial composition of D. fragilis cultures and sequence the D. fragilis 28S rDNA. While the persistence of bacterial reads within sequence data has proven a difficult hurdle to overcome, a significant amount of D. fragilis related reads are present within the source material as shown by the mapping of over 800 000 reads to trichomonad related reference indexes. We continue to investigate alternative bioinformatics approaches for the generation of D. fragilis genome data and remain optimistic that meaningful D. fragilis genomic data can be generated from this methodology. Ultimately, however, increasing the number of D. fragilis trophozoites in the culture still remains a hurdle to overcome as it remains a limiting factor in the sequencing strategy, even with the inclusion of K. pneumoniae in the cell culture.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182019001173
Financial support
This research received no specific grant from any funding agency, commercial or not-for-profit sectors. R. Gough received a PhD scholarship from the University of Technology Sydney. J. Barratt received a Chancellor's postdoctoral fellowship from UTS.
Conflict of interest
None.
Ethical standards
Not applicable.








