


Ellis – van Creveld syndrome (also known as ectodermal dysplasia, Online Mendelian Inheritance in Man 225,500) is an autosomal recessive disorder that was first described in 1940.Reference Ellis and van Creveld1 Clinical manifestations of Ellis – van Creveld syndrome include short-limb dwarfism, postaxial polydactyly, abnormalities in tooth and nail development, congenital malformations of the heart, thoracic dystrophy, Dandy–Walker malformations, and hepatic abnormalities.Reference McKusick, Egeland, Eldridge and Krusen2–Reference Abeles and Tobias4 Cognitive development is normal. Despite its rarity in the general population (approximately 1 in 20,000 births), Ellis – van Creveld syndrome is common among the Old Order Amish of Lancaster County, Pennsylvania, where the carrier frequency may be as high as 13% and the disease incidence is 0.5%.Reference McKusick, Egeland, Eldridge and Krusen2, Reference Baujat and Le Merrer3, Reference McKusick5 Ellis – van Creveld syndrome is caused by loss-of-function mutations in either the EVC or EVC2 genes located on chromosome 4p16.Reference Ruiz-Perez, Ide and Strom6, Reference Ruiz-Perez, Tompson and Blair7 The subcellular localisation and function of EVC has recently been elucidated,Reference Ruiz-Perez, Blair and Rodriguez-Andres8, Reference Ruiz-Perez and Goodship9 leading to an emerging understanding of how EVC mutations cause the clinical phenotype.
Approximately 60% of patients with Ellis – van Creveld syndrome have congenital malformations of the heart.Reference McKusick, Egeland, Eldridge and Krusen2, Reference Digilio, Marino, Ammirati, Borzaga, Giannotti and Dallapiccola10 The spectrum of disease is broad, but most commonly includes defects in atrial and ventricular septation, particularly atrioventricular septal defect with a common atrium. However, complex lesions such as hypoplastic left heart syndrome and heterotaxy syndrome can occur; in fact, the types of congenital malformations of the heart encountered in Ellis – van Creveld syndrome closely resemble those seen in the left atrial isomerism form of heterotaxy syndrome (Figs 1 and 2).Reference Digilio, Marino, Ammirati, Borzaga, Giannotti and Dallapiccola10 Ellis – van Creveld syndrome is only one of several syndromes known to be associated with atrioventricular septal defects. The distinctive manifestations of atrioventricular septal defects in patients without trisomy 21 have been investigated in detail. For example, systemic ventricular outflow tract obstruction in the setting of atrioventricular septal defects appears to be more common in syndromic or non-syndromic patients without trisomy 21.Reference Digilio, Marino, Toscano, Giannotti and Dallapiccola11, Reference De Biase, Di Ciommo, Ballerini, Bevilacqua, Marcelletti and Marino12 The most comprehensive clinical description of Ellis – van Creveld syndrome was published by McKusick and colleagues in 1964, prior to the widespread availability of surgery for congenital malformations of the heart.Reference McKusick, Egeland, Eldridge and Krusen2 In that series of 52 patients, mortality of 58% in infants less than 6 months of age was attributed to both congenital malformations of the heart and malformations of the lungs and chest wall. In contrast, individuals without congenital malformations of the heart had normal survival.Reference McKusick, Egeland, Eldridge and Krusen2, Reference Ruiz-Perez and Goodship9 Through this early work, it became evident that cardiopulmonary features of the syndrome are the main determinants of survival. It is presently unclear, however, whether progress in the care of children with complex congenital malformations of the heart has similarly resulted in improved clinical outcomes for patients with Ellis – van Creveld syndrome and congenital malformations of the heart. To address this question, we studied nine children with Ellis – van Creveld syndrome who underwent surgery for congenital malformations of the heart during the recent era.

Figure 1 A two-dimensional apical four-chamber echocardiographic image in a 1-day-old infant with Ellis – van Creveld syndrome. (a) a common atrioventricular valve in diastole; (b) a common atrioventricular valve in systole. There is a complete atrioventricular septal defect with a common atrium (CA). The common atrioventricular valve is delineated by asterisks. There is significant size discrepancy between the right ventricle (RV) and left ventricle (LV), indicating an imbalance of the common atrioventricular septal defect at the ventricular level (favouring the RV).

Figure 2 Two-dimensional suprasternal sagittal echocardiographic image of the same patient displayed in Fig 1. Note the hypoplasia of the transverse aorta (asterisk) and aortic isthmus (arrowhead). The diameter of the transverse aorta is 0.25 centimetre (z score −3.6) and the diameter of the aortic isthmus is 0.25 centimetre (z score −3.9). The ascending aorta is of normal size.
Materials and methods
Institutional review board approval was obtained from The Children’s Hospital of Philadelphia. Patients with Ellis – van Creveld syndrome born between 1 January, 2004 and 31 December, 2009 were identified retrospectively using clinical databases at either the Clinic for Special Children or The Children’s Hospital of Philadelphia. The diagnosis of Ellis – van Creveld syndrome was established by a clinical geneticist, after exclusion of phenotypically related conditions. Patients who underwent surgery for congenital malformations of the heart during infancy comprised the cohort described within this report; those patients without congenital malformations of the heart or whose defects were so trivial as to not warrant surgery (for example, patent oval foramen) were excluded from analysis. Surgery for congenital malformations of the heart was performed at one of three regional paediatric cardiac surgical centres. Cardiac diagnoses were determined by a review of echocardiogram and cardiac catheterisation data and confirmed by a review of operative reports.
Patients were categorised as having pulmonary hypertension if, at cardiac catheterisation, mean pulmonary artery pressure was greater than 25 millimetres of mercury with a normal pulmonary capillary wedge pressure, and/or pulmonary vascular resistance was greater than 3 indexed Wood units.Reference McLaughlin, Archer and Badesch13 Echocardiographic parameters such as right ventricular systolic pressure estimate through tricuspid valve regurgitant jet velocity and interventricular septal position were considered suggestive of pulmonary hypertension but were not used for diagnostic confirmation. Follow-up on all patients was obtained through 1 May, 2010.
Results
Patients and demographics
A total of 15 consecutive patients with Ellis – van Creveld syndrome, all born between 1 January, 2004 and 31 December, 2009, presented for care at either The Children’s Hospital of Philadelphia or The Clinic for Special Children. All underwent screening for congenital malformations of the heart via echocardiographic examination. Of the 15 patients, 11 (73%) had haemodynamically significant congenital malformations of the heart deemed appropriate for surgical correction and/or palliation (Fig 3); two of these were children (both with complete common atrioventricular septal defects) who did not undergo surgery; one had neonatal respiratory failure and died at 5 days of age; the other had multiple coexisting congenital abnormalities and was lost to follow-up. Thus, nine infants presented for surgery during the time period of this report, of whom eight were Amish.

Figure 3 Flow diagram of the patients identified with Ellis – van Creveld syndrome in this series.
Pre-operative respiratory failure requiring mechanical ventilation of any duration was present in five of the nine (56%) patients. The cardiac diagnoses, surgeries performed, and additional relevant clinical information are presented in Table 1. The median age at surgery was 66 days with a range from 2 to 144 days, with a median weight at surgery of 3.8 kilograms ranging from 2 to 4.7 kilograms. The median duration of post-operative follow-up was 0.8 years with a range from 25 days to 5.9 years.
Table 1 Clinical characteristics of the patients.

ASD = atrial septal defect; AV = atrioventricular; AVSD = atrioventricular septal defect; CS = coronary sinus; HLHS = hypoplastic left heart syndrome; LA = left atrium; LSCV = left superior caval vein; NA = data not available; RA = right atrium; RSV = respiratory syncytial virus
*Present pre- and post-operatively
**Confirmed at cardiac catheterization
***Following hemi-Fontan procedure
****Placed prior to cardiac surgery
Survival
Of the nine patients, four (44%) died after surgery, at a median of 102 days with a range from 25 to 149 days post-operatively (Table 1). Patient number 2 died 113 days post-operatively, from respiratory syncytial virus bronchiolitis, hypercarbic respiratory failure, and hypoxemia refractory to aggressive ventilatory support. Patient number 3 was diagnosed with a transitional atrioventricular septal defect at 1 month of age. By 2 months of age, this patient presented with severe hypercarbic respiratory failure and echocardiographic findings suggestive of pulmonary hypertension. Repair of the atrioventricular septal defect was performed, but the patient died 3 months later after the family declined tracheostomy. Patient number 7 underwent repair of the common atrium and a transitional atrioventricular septal defect at 3 weeks of age. This child’s medical history was complicated by prematurity and a Dandy–Walker malformation that required placement of a ventriculoperitoneal shunt to manage hydrocephalus. The patient could not be weaned from mechanical ventilation following repair of the cardiac defect, and support was withdrawn 25 days post-operatively. Patient number 8 died 4 months following surgery, from a cardiac arrest of unknown aetiology.
Discussion
Management of congenital malformations of the heart in Ellis – van Creveld syndrome is associated with substantial mortality. In particular, the respiratory system appears to be central with respect to post-operative outcome. This is in keeping with the concept that syndrome-specific clinical manifestations may play a significant role in both the management and outcome of surgery for congenital diseases of the heart.Reference Formigari, Di Donato and Gargiulo14, Reference Formigari, Michielon and Digilio15 In this series, nearly half of the patients who underwent surgery died, and respiratory morbidity was encountered in all survivors. Respiratory failure was the cause of death in all but one case, and further resulted in post-operative tracheostomy in three surviving patients. In comparison, we are unaware of any mortality in those patients with Ellis – van Creveld syndrome without congenital malformations of the heart (four patients) born during the time frame of this study.
The severity of respiratory disease varied along a broad spectrum that included persistent post-operative tachypnea, hypoxaemia requiring nasal cannula oxygen, and fatal respiratory failure. Prolonged mechanical ventilation was often required (see Table 1), and three patients underwent tracheostomy. Aside from the baseline thoracic dystrophy, clinical predictors of respiratory failure in patients with Ellis – van Creveld syndrome remain unknown. A restrictive chest wall, diminished lung capacity, and general debilitation most likely rendered post-operative patients more vulnerable to respiratory infections, and respiratory syncytial virus infection proved fatal in one case. Median sternotomy in patients with Ellis – van Creveld syndrome may also impair respiratory mechanics. This has been reported after early childhood repair of pectus excavatum deformities, which can result in an iatrogenic restrictive lung disease, termed “acquired Jeune syndrome.”Reference Haller, Colombani, Humphries, Azizkhan and Loughlin16, Reference Robicsek and Fokin17
As initially described by McKusick,Reference McKusick, Egeland, Eldridge and Krusen2 neonatal respiratory failure, mediated in part by abnormalities of the thoracic cage, is a prominent feature of Ellis – van Creveld syndrome (Fig 4). The chest wall physiology in Ellis – van Creveld syndrome resembles that of Jeune syndrome, also known as asphyxiating thoracic dystrophy. Both are associated with polydactyly, short ribs, and an increased risk of restrictive lung disease and respiratory failure.Reference Phillips, Stokoe and Bartholomew18–Reference Krakow, Salazar, Wilcox, Rimoin and Cohn22 The pathophysiology of these syndromes may converge at the cellular level. Recent work has shown that EVC and EVC2 are localised to the base of primary cilia in developing tissues, where they act as downstream elements of the hedgehog signalling pathway.Reference Ruiz-Perez, Blair and Rodriguez-Andres8 Hedgehog signalling affects morphogenesis of the heart, lung, and pulmonary vasculature, and indicates a more appropriate designation for Ellis – van Creveld syndrome, proposed over 50 years ago: mesoectodermal dysplasia.Reference Mitchell and Waddell23 Mutations in separate cilia-related proteins have recently been reported in patients with Jeune syndrome.Reference Ruiz-Perez and Goodship9, Reference Dagoneau, Goulet and Genevieve24 Thus, defective hedgehog signalling within primary cilia may provide a unifying explanation for the protean manifestations of short rib-polydactyly syndromes, as has been suggested by Digilio et al.Reference Digilio, Marino, Giannotti, Dallapiccola and Opitz25
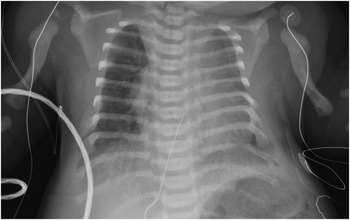
Figure 4 Anteroposterior portable chest radiograph in a newborn infant with Ellis – van Creveld syndrome, common atrium, and an unbalanced atrioventricular septal defect demonstrating the thoracic dystrophy acccompanying Ellis – van Creveld syndrome. There is cardiomegaly with increased pulmonary vascular markings. The chest is bell-shaped, with short ribs. The humeri are also short.
The clinical course of Ellis – van Creveld syndrome can resemble that of Jeune syndrome. In a recent series of 11 patients with Jeune syndrome, mortality was 54%; 83% of these deaths were attributed to respiratory failure.Reference Tüysüz, Baris, Aksoy, Madazli, Üngür and Sever26 In Jeune syndrome patients, respiratory function may improve after 2 years of age,Reference de Vries, Yntema, van Die, Crama, Cornelissen and Hamel27 and our clinical observations suggest that growth also stabilises respiratory function in Ellis – van Creveld syndrome. Accordingly, it may be reasonable to defer cardiac surgery beyond infancy when possible in patients with Ellis – van Creveld syndrome who are not dependent upon a patent arterial duct for systemic or pulmonary blood flow. However, such a strategy must be balanced against the risks associated with unrepaired congenital malformations of the heart, including chronic cyanosis, failure to thrive, and pulmonary vascular disease. Thus, the optimal timing of surgery in many patients with Ellis – van Creveld syndrome and congenital malformations of the heart remains far from clear.
The risk for pulmonary hypertension in patients with Ellis – van Creveld syndrome complicates medical decision-making. Direct or indirect evidence of pulmonary hypertension was observed in nearly half of the cohort, and three of those patients died. We could not determine the true incidence of pulmonary hypertension in our cohort, as all patients did not undergo cardiac catheterisation. Pulmonary vascular disease should be considered in any patient with Ellis – van Creveld syndrome and a large left-to-right intracardiac shunt, such as common atrium or atrioventricular septal defect. Classical teaching dictates that the common atrium is associated with accelerated development of pulmonary vascular disease, and thus warrants early surgical repair.Reference Cetta, Minich, Edwards, Dearani and Puga28 In patients with Ellis – van Creveld syndrome, pulmonary vascular disease may result from the interplay of several physiologic variables, including restrictive lung mechanics, diminished lung volumes, intercurrent respiratory infections, increased pulmonary blood flow, and chronic cyanosis from intercirculatory mixing. Of note, hedgehog signalling may directly influence development of the pulmonary vasculature,Reference White, Lavine and Ornitz29 and disturbances in this pathway may partly explain the occurrence of pulmonary vascular disease in some patients with Ellis – van Creveld syndrome. Whatever the cause, the risk for pulmonary vascular disease persists despite a technically successful surgery. Cardiac catheterisation to evaluate pulmonary vascular pressures and resistance should be considered in any post-operative patient with cardiopulmonary failure, and may be indicated pre-operatively to determine suitability for surgery.
Complex cardiac disease is seen in patients with Ellis – van Creveld syndrome, as evidenced by the cardiac diagnoses observed in this and previous series. It should be noted that persistent left superior caval vein draining to an intact or unroofed coronary sinus was encountered in one-third of the patients. The association of this finding with a common atrium has been noted in prior studiesReference Digilio, Marino, Ammirati, Borzaga, Giannotti and Dallapiccola10; therefore, careful pre-operative delineation of the systemic venous anatomy is crucial for the purposes of selecting the correct surgical approach.
In this series, two patients were committed to single ventricle palliation for variations of hypoplastic left heart syndrome. To our knowledge, the outcomes of this approach in Ellis – van Creveld syndrome have not been described in the literature. In the setting of impaired respiratory mechanics, the strategy of single ventricle palliation warrants special mention. Single ventricle palliation requires a series of surgeries that culminate in the Fontan procedure, which allows for separation of the circulation without a ventricle being interposed between the systemic and pulmonary venous beds. The importance of a low-pressure gradient through the Fontan pathway to successful single ventricle palliation has been appreciated since the 1970s.Reference Hosein, Clarke and McGuirk30 Despite the impaired respiratory mechanics in Ellis – van Creveld syndrome in principle compromising the surgical approach to single ventricle palliation, the two patients with Ellis – van Creveld syndrome in this report who have successfully been palliated along this pathway have had reasonable clinical outcomes.
Limitations
Despite an increased incidence among the Amish, Ellis – van Creveld syndrome remains rare, making it difficult to study large numbers of patients during a period of relatively uniform clinical practice standards for congenital malformations of the heart. This limits our ability to generalise our observations. Despite the thoracic dystrophy accompanying Ellis – van Creveld syndrome appearing to play a substantial role in the outcomes of patients in this series, the patients in this series did not undergo formalised assessments of respiratory function pre-operatively. In clinical practice, thoracic dystrophy, particularly in infants, remains difficult to assess in a standardised manner. It is apparent, therefore, that a more rigorous approach to pre-operative assessment of respiratory function and capacity is necessary in this patient population. By focusing on infants who underwent repair of complex defects, we recognise that less complex forms of congenital malformations of the heart (for example, small atrial septal defects) may be underrepresented, since such defects frequently do not come to clinical attention until later in childhood. In this group of patients, the clinical course may be distinctly different. Infants, however, appear to experience the highest rates of mortality in Ellis – van Creveld syndrome; therefore, their clinical outcomes are of significant interest to both families and clinicians, given that they represent the group of patients at highest risk of morbidity and mortality.
Conclusion
Surgical repair or palliation of complex congenital malformations of the heart in patients with Ellis – van Creveld syndrome can be successful, but overall mortality remains high in the current era (44%) and post-operative respiratory morbidity should be expected. Aggressive ventilatory management, including tracheostomy, may be necessary to compensate for underlying thoracic dystrophy. The complex nature of the thoracic cage and lung abnormalities in Ellis – van Creveld syndrome may be dynamic and not readily apparent before cardiac surgery. In this series, some infants who appeared to be good candidates fared poorly after surgery. This underscores the need for careful pre-operative evaluation of respiratory mechanics, ventilatory function, and pulmonary vascular status, but does not ensure that such evaluation will predict a satisfactory post-operative outcome. Future research should focus on the complex interactions among chest wall mechanics, pulmonary vascular function, and cardiac physiology in Ellis – van Creveld syndrome. A deeper understanding of fundamental cellular derangements in Ellis – van Creveld syndrome and their consequences with respect to cardiopulmonary physiology will be crucial for improving the health and survival of affected children.
Acknowledgements
Financial Disclosures: None
Conflicts of Interest: None





