


INTRODUCTION
As a disease of poverty, schistosomiasis has proven difficult to control for centuries, and continues to have a significant clinical and socioeconomic impact in terms of human morbidity and mortality (WHO, 2008, 2010; Murray et al. Reference Murray, Barber, Foreman, Abbasoglu Ozgoren, Abd-Allah, Abera, Aboyans, Abraham, Abubakar, Abu-Raddad, Abu-Rmeileh, Achoki, Ackerman, Ademi, Adou, Adsuar, Afshin, Agardh, Alam, Alasfoor, Albittar, Alegretti, Alemu, Alfonso-Cristancho, Alhabib, Ali, Alla, Allebeck, Almazroa and Alsharif2015). It is estimated that schistosomiasis causes ~41 000 deaths annually with ~700 million people at risk of infection in tropical and sub-tropical areas (Murray et al. Reference Murray, Barber, Foreman, Abbasoglu Ozgoren, Abd-Allah, Abera, Aboyans, Abraham, Abubakar, Abu-Raddad, Abu-Rmeileh, Achoki, Ackerman, Ademi, Adou, Adsuar, Afshin, Agardh, Alam, Alasfoor, Albittar, Alegretti, Alemu, Alfonso-Cristancho, Alhabib, Ali, Alla, Allebeck, Almazroa and Alsharif2015). Furthermore, with increased international travel and global immigration, the disease is frequently reported in returning travellers and in migrants, and as such reports of its emergence into temperate localities, including Europe and the USA are increasing (Ross et al. Reference Ross, Olds, Cripps, Farrar and McManus2013; Boissier et al. Reference Boissier, Moné, Mitta, Bargues, Molyneux and Mas-Coma2015; Gautret et al. Reference Gautret, Mockenhaupt, Bottieau, Parola and Schlagenhauf2015; Gordon et al. Reference Gordon, McManus, Jones, Gray and Gobert2016). Schistosomiasis japonica, caused by Schistosoma japonicum, is a zoonosis, and in China and the Philippines it is present in water buffaloes and other bovines as major reservoirs for human infection, which complicates efforts for disease control and elimination (Gordon et al. Reference Gordon, Acosta, Gray, Olveda, Jarilla, Gobert, Ross and McManus2012, Reference Gordon, Acosta, Gobert, Olveda, Ross, Williams, Gray, Harn, Li and McManus2015a ). The World Health Organization (WHO) strategic plan for 2012–2020 (WHO, 2013) calls for the elimination of schistosomiasis as a public health problem by the year 2025. To this end a number of prevention and control programmes are currently underway in efforts to reach this goal. These are chiefly focused on mass drug administration or targeted treatment of infected patients with the drug praziquantel. Schistosomiasis prevalence and infection intensity in endemic areas may therefore be reduced following regular treatment regimens with this drug. However, elimination requires confirmation that transmission has ended and no new cases are appearing. Consequently, it is important to have available sensitive diagnostic tools that can detect low prevalence and low-intensity infections, particularly within the framework of population surveillance and the monitoring of schistosomiasis control programmes. Furthermore, the acute form of schistosomiasis is not diagnosed early due to the lack of diagnostic tools that are sufficiently sensitive to detect schistosome parasites during the first weeks of infection (Sandoval et al. Reference Sandoval, Siles-Lucas, Lopez Aban, Pérez-Arellano, Gárate and Muro2006).
Of the different diagnostic procedures currently available, conventional parasitological methods, such as the Kato-Katz (KK) thick smear stool examination have the major limitation of low diagnostic accuracy, particularly in areas with low transmission (Kongs et al. Reference Kongs, Marks, Verlé and Van der Stuyft2001; Spear et al. Reference Spear, Seto, Carlton, Liang, Remais, Zhong and Qiu2011; Weerakoon et al. Reference Weerakoon, Gobert, Cai and McManus2015). Serological techniques also lack the required sensitivity and specificity in low-intensity settings (Weerakoon et al. Reference Weerakoon, Gobert, Cai and McManus2015). Circulating antigen-based detection assays provide improved accuracy compared with antibody detection assays, but day-to-day fluctuations in assay results suggest the need for multiple sample testing to increase diagnostic sensitivity (Stothard et al. Reference Stothard, Kabatereine, Tukahebwa, Kazibwe, Rollinson, Mathieson, Webster and Fenwick2006; Legesse and Erko, Reference Legesse and Erko2007). Recently, monoclonal antibody-based antigen detection methods, especially a specialized technique, the UCP-LF (upconverting phosphor lateral flow assay), have proven successful with high sensitivity to detect CAA (circulating anodic antigen) in the serum and urine of subjects with schistosomiasis mansoni; however, only limited testing has been undertaken to detect individuals with S. japonicum (Corstjens et al. Reference Corstjens, De Dood, Kornelis, Fat, Wilson, Kariuki, Nyakundi, Loverde, Abrams, Tanke, Van Lieshout, Deelder and Van Dam2014; Utzinger et al. Reference Utzinger, Becker, van Lieshou, van Dam and Knopp2015; van Dam et al. Reference van Dam, Xu, Bergquist, de Dood, Utzinger, Qin, Guan, Feng, Yu, Zhou, Zheng, Zhou and Corstjens2015).
It is therefore necessary to develop more patient-friendly, convenient and accurate diagnostic tools to support schistosomiasis elimination programmes as well as for detection of pre-patent schistosome infections. DNA-based detection methods provide a viable alternative to some of the commonly used tests for the diagnosis of schistosomiasis japonica (Xu et al. Reference Xu, Rong, Zhang, Shi, Zhu and Xia2010; Gordon et al. Reference Gordon, Gray, Gobert and McManus2011, Reference Gordon, Acosta, Gobert, Olveda, Ross, Williams, Gray, Harn, Li and McManus2015a ). The detection of parasite cell-free DNA (cfDNA) in different clinical samples is a recent valuable advance, which provides significant benefits towards the accurate early diagnosis of schistosomiasis (Kato-Hayashi et al. Reference Kato-Hayashi, Yasuda, Yuasa, Isaka, Haruki, Ohmae, Osada, Kanazawa and Chigusa2013, Reference Kato-Hayashi, Leonardo, Arevalo, Tagum, Apin, Agsolid, Chua, Villacorte, Kirinoki, Kikuchi, Ohmae, Haruki and Chigusa2015; Weerakoon and McManus, Reference Weerakoon and McManus2016). cfDNA is comprised fragments of extracellular DNA found mainly in the blood circulation, but it can also be detected in other bodily fluids such as saliva and urine. Parasite cfDNA is released into the circulation, originating from different stages of the schistosome parasite, and is uniformly distributed in plasma, unlike schistosome eggs in feces or urine. Detection of cfDNA may overcome sampling errors, a major limitation, for example of egg DNA detection in stool samples. To date, the droplet digital polymerase chain reaction (ddPCR) has exhibited the highest diagnostic accuracy for DNA detection, and is able to provide absolute quantification of target gene copy numbers in a sample. ddPCR is an endpoint quantitative PCR (qPCR) and, compared with real-time qPCR-based assays, it is more accurate and sensitive in different clinical applications such as pathogen detection, cancer diagnosis and prenatal testing (Miotke et al. Reference Miotke, Lau, Rumma and Ji2014; Sze et al. Reference Sze, Abbasi, Hogg and Sin2014; Hudecova, Reference Hudecova2015; Weerakoon et al. Reference Weerakoon, Gordon, Gobert, Cai and McManus2016). Here we describe the validation of a novel duplex ddPCR assay we developed (Weerakoon et al. Reference Weerakoon, Gordon, Gobert, Cai and McManus2016) for the diagnosis of Chinese (SjC) and Philippine (SjP) strains of S. japonicum infection in a mouse model, as a precursor for its field application.
MATERIALS AND METHODS
Ethics statement
This study was conducted with the approval (approval numbers P288, P443) of the QIMR Berghofer Medical Research Institute (QIMRB) animal and human ethics committees, and was performed in accordance with the Australian code of practice for the care and use of animals for scientific purposes (7th edition; 2004).
Parasites
Schistosoma japonicum cercariae from China (Guichi County, Anhui) and the Philippines (Irosin, near Sorsogon, Southern Luzon) were shed from infected Oncomelania hupensis hupensis and O. hupensis quadrasi snails, respectively. Mice were then percutaneously infected using shed cercariae. The parasite life cycles are maintained routinely in the laboratory at QIMRB using infected snails originally collected from the field.
Establishment of S. japonicum-infections in mice and sample collection
Twelve female ARC Swiss mice were subjected to transcutaneous cercarial infection. Mice were divided into two groups (A and B) of six. Group A was infected with SjP (70 cercariae/mouse) and Group B was infected with SjC (50 cercariae/mouse) (Fig. 1). A higher challenge dose of cercariae was used in the experimental infection of mice with SjP than SjC due to the known higher virulence of the latter (Hope et al. Reference Hope, Duke and McManus1996). Blood, urine and stool samples were collected at four different time points – pre-infection controls and 1, 3 and 6 weeks post-infection; mice were sacrificed immediately after the last time point. Blood samples (~150 µL) were collected by tail vein puncture; serum was separated by centrifuging the clotted blood samples at 1500 g for 10 min. Serum was frozen at −80 °C until used for DNA extraction. The remaining blood clot was digested (following the protocol described below under tissue digestion and egg count analysis) to test for the presence of eggs. This step was performed to check for the presence or absence of eggs in these samples to confirm that the assay was detecting cfDNA and not DNA from eggs. Urine samples were centrifuged at 1500 g for 10 min and the supernatant stored at −80 °C until DNA was extracted. The urine sediment was microscopically examined for the presence of schistosome eggs. Stool samples were stored in 4 °C after fixing in 80% (v/v) ethanol until DNA extraction and egg count analysis. Worm counts were obtained at the time of sacrifice and perfusion of mice. Liver and intestinal samples were also collected for S. japonicum egg counts. In addition, salivary glands and urinary bladders were collected from a third group of six female ARC Swiss mice infected with SjP (70 cercariae/mouse) at 6 weeks post-infection and the infection status of these was determined by worm and fecal and hepatic egg counts. Salivary glands were washed with phosphate-buffered saline (PBS) and stored at −80 °C for subsequent DNA extraction and PCR analysis. Urinary bladders were stored in 4 °C after fixing in 80% ethanol to test for the presence of eggs by tissue digestion. This was done to confirm that the S. japonicum DNA detected in urine was due to cfDNA and not to the presence of parasite eggs.

Fig. 1. Time line and overview of the animal experiments.
Cercarial transformation, maintenance of schistosomula and adult worm cultures, and sample collection
SjP adult worms and schistosomula were cultured in vitro. Culture supernatants were collected to test for the presence of schistosome DNA. Mechanical transformation of cercariae to schistosomula, and their subsequent in vitro cultivation, was performed as described previously (Milligan and Jolly, Reference Milligan and Jolly2011). Four groups of 100 schistosomula were cultivated separately in a 96-well U bottom plate with 200 µL of Basch medium (Basch, Reference Basch1981) with 1:100 human red blood cells, 0·1 U µL−1 penicillin and 0·1 µg µL−1 streptomycin, in an incubator with an atmosphere of 5% CO2 in air. The culture supernatant from each of the four groups was collected daily for five consecutive days by centrifuging individual plates at 180 g for 10 min and placing the supernatant into a microcentrifuge tube, without disturbing the pellet of live schistosomula. Wells were then replaced with another 200 µL of new culture media. The culture supernatant was further centrifuged at 14 000 g for 10 min and stored at −80 °C for subsequent DNA extraction and PCR analysis.
Schistosoma japonicum adult worms were collected by perfusion of SjP cercariae-infected female ARC Swiss mice at day 42 post-infection. Adult worms were then incubated in the sterile RPMI-1640 medium with 20% (v/v) fetal bovine serum, 100 U mL−1 penicillin, and 100 µg mL−1 streptomycin, in an incubator with an atmosphere of 5% CO2 in air (You et al. Reference You, Zhang, Jones, Gobert, Mulvenna, Rees, Spanevello, Blair, Duke, Brehm and McManus2010). Four groups of worms, each comprising five worm pairs, were cultivated in 1·5 mL of culture medium. The culture supernatant was collected daily for 5 days as described above for the culture of schistosomula. Following each collection, fresh culture medium was added to each plate. The collected culture supernatant was then centrifuged at 14 000 g for 10 min and stored at −80 °C for subsequent DNA extraction and PCR analysis.
Harvesting and counting of eggs
Livers from infected mice were weighed, chopped into small pieces, placed into 50 mL conical centrifuge tubes and digested in 10 mL 4% (w/v) KOH at 37 °C overnight. The digested liver extracts were then centrifuged at 2000 g for 10 min, the sediment was resuspended in 5 mL of 4% KOH, and the procedure and centrifugation repeated. The resulting pellet was then resuspended in 5 mL of 5% (v/v) buffered formalin and examined microscopically. Three 10 µL aliquots from each sample were examined for eggs and the mean egg count was used to calculate eggs per gram (EPG) of feces. The identical procedure was followed for the examination of eggs in the intestine, bladder and blood clots.
Fecal samples were processed by adding 5 mL of 10% (v/v) neutral buffered (PBS) formalin to weighed samples in 10 mL conical tubes, which were placed in a rotating wheel overnight at 4 °C. The resulting stool suspensions were then homogenized and passed through a 200 µm aperture nylon cloth, which was then rinsed into 10 mL conical tubes with 3% (v/v) formalin in PBS. The tubes were centrifuged at 3000 rpm for 10 min and the resulting sediment was subjected to KOH digestion as described above.
DNA extraction
DNA was isolated from the feces, urine, serum and salivary gland samples collected from mice at different time points. QIAmp DNA stool mini kits, QIAmp DNA Blood mini kits and QIAmp viral RNA mini kits (Qiagen, Hilden, Germany) were used for the extraction of DNA from feces, serum and urine, respectively, following the manufacturer's guidelines. DNA extraction from the salivary glands and adult S. japonicum worm and schistosomula culture supernatants was undertaken using DNeasy blood and tissue kits (Qiagen, Hilden, Germany). DNA quantification and quality were tested using a Nanodrop 1000 (Thermo Scientific, Waltham, USA) spectrophotometer. Subsequent PCR analysis was undertaken using DNA concentrations of 50 ng µL−1 for fecal, urine and salivary gland samples and 5 ng µL−1 concentrations for serum and culture supernatant DNA samples.
PCR assays
The mitochondrial NADH dehydrogenase 1 (nad1) and the SjR2 retrotransposon were selected as target genes, as these sequences are highly abundant in the S. japonicum genome (Lier et al. Reference Lier, Simonsen, Haaheim, Hjelmevoll, Vennervald and Johansen2006; Schistosoma japonicum Genome Sequencing and Functional Analysis Consortium, 2009). The primers employed for the PCR assay amplified a 82 bp fragment of the nad1 gene (GenBank Accession No: NC002544) (Lier et al. Reference Lier, Simonsen, Haaheim, Hjelmevoll, Vennervald and Johansen2006; Gordon et al. Reference Gordon, Acosta, Gray, Olveda, Jarilla, Gobert, Ross and McManus2012; Weerakoon et al. Reference Weerakoon, Gordon, Gobert, Cai and McManus2016): 5′-TGRTTTAGATGATTTGGGTGTGC-3′ (Forward) and 5′-AACCCCCACAGTCACTAGCATAA-3′ (Reverse), and a 191 bp fragment of the highly repetitive SjR2 gene (GenBank Accession No: AY027869·1) (Weerakoon et al. Reference Weerakoon, Gordon, Gobert, Cai and McManus2016), 5′-CAGTGAAGTTGTGAAGGCTA-3′ (Forward) and 5′-TAAAGACCGGAATGGTTAGT-3′ (Reverse).
A novel duplex digital PCR assay (Weerakoon et al. Reference Weerakoon, Gordon, Gobert, Cai and McManus2016) was employed in the analysis of the different sample types. Reaction mixtures of 20 µL were used comprising 10 µL 1× ddPCR EvaGreen master mix (Bio-Rad, Hercules, USA), 100 nm (final concentration) of each of the SjR2 forward and reverse primers, 50 nm (final concentration) of each of the nad1 forward and reverse primers, 2 µL of template DNA and 4 µL of distilled H2O. The reaction mix was pipetted into the sample well of aneight channel disposable droplet generator cartridge (BioRad). For each cartridge, 70 µL of droplet generation oil for EvaGreen (BioRad) was loaded into the oil wells, covered with a rubber gasket and then placed into a droplet generator (Bio-Rad). The generation of droplets resulted in partitioning the 20 µL sample into ~20 000 nL-sized droplets and the droplets were transferred to a 96-well twin.tec™ PCR plate (Eppendorf, Hamburg, Germany). The twin.tec plate was then heat sealed with a pierceable foil heat seal (BioRad) using a PX1 PCR Plate Sealer (BioRad) and placed into a thermal cycler (c1000, BioRad). The two-step ddPCR cycling conditions were as follows: an initial enzyme activation period of 5 min at 95 °C, 40 cycles of denaturation at 95 °C for 30 s, and annealing/extension temperature of 55·8 °C for 1 min, followed by a single final dye stabilization step of 4 °C for 5 min and then 95 °C for 5 min. Negative controls (no DNA template controls) were used in all the assays, as were positive controls containing DNA extracted from adult worms. Following the PCR amplification, the plate was placed in a QX200 Droplet Reader (BioRad) for analysis. Droplets from each well were automatically aspirated and sent through a two colour fluorescent detector to determine the fluorescent amplitude of each well. Droplet analysis was performed using Quanta-soft (BioRad) software.
Conventional PCR (cPCR) was used to amplify the SjR2 retrotransposon and nad1 gene (Weerakoon et al. Reference Weerakoon, Gordon, Gobert, Cai and McManus2016) to further confirm the target product amplification size. Reaction mixtures were prepared containing 0·125 µL of MyTaq DNA polymerase (Bioline, London, UK), 0·5 µL of each primer from an initial 10 µ m solution (250 nm final conc.) (Sigma-Aldrich, St. Louis, USA), 5 µL of MyTaq PCR Buffer mix (Bioline), 2 µL of template DNA and distilled H2O to a final volume of 25 µL. The following cycling conditions were used for the cPCR with SjR2: initial denaturation at 95 °C for 2 min, followed by 35 cycles of 95 °C (15 s), annealing at 55 °C (15 s), extension at 72 °C (15 s) and a final extension at 72 °C for 2 min. Similar cycling conditions were used with the nad1 primers, except for the annealing temperature which was increased to 59 °C for 20 s. As for the ddPCR, negative (no DNA template) and positive (adult worm DNA) controls were used in all assays. The PCR products were analysed visually after electrophoresis through 2% (w/v) agarose gels stained with SYBR® Safe DNA Gel Stain (Invitrogen, Carlsbad, USA). A 100 bp ladder was added to the gel and the amplicons checked for the expected band size.
Statistical analysis
GraphPad Prism 6·05 (GraphPad Software, Inc., La Jolla, USA) and Microsoft Excel were used for all statistical analyses. Results are expressed as means and standard errors of the mean (s.e.m.) when presenting summary measures. Two sample t-test was used to compare differences among the infection burdens of the two mouse groups. Correlations between DNA quantities and infection burden at different infection time points were tested with Pearson's correlation analysis, after using the transformation log (x + 1), where ‘x’ represents the DNA copy number or EPG.
RESULTS
Parasitological detection of S. japonicum infection in mouse groups
The infection status of the two mouse groups (A and B) were confirmed with the detection of schistosome eggs in fecal, intestinal and liver samples, and adult worm pair counts obtained following perfusion at day 42 (Table 1). All egg counts were significantly higher in the SjC infected mouse group compared with the SjP group (P < 0·05, two sample t-test). Fecal samples collected at days 7 and 21 post-infection were negative for schistosome eggs. Eggs were not found in urine sediments, digested blood clots (following serum separation), or urinary bladders collected at perfusion on day 42.
Table 1. Infection status of the SjP (group A)- and SjC (group B)-challenged mice

EPG, Eggs per gram; s.e.m., standard error of mean.
ddPCR analysis of mouse samples
The duplex ddPCR assay detected both SjR2 and nad1 target sequences in fecal, urine and serum samples collected on days 7, 21 and 42 post-infection (Fig. 2). Pre-infection controls of all sample types were negative in all 12 mice. Quantitatively the highest gene copy numbers were detected at day 42 for all three sample types, in both the SjP and SjC groups. However, the gene copy numbers at day 7 and day 21 showed high variability between sample type, day and parasite strain (Fig. 2). The correlation analysis confirmed high correlations between infection intensity (fecal, liver an intestinal egg densities) and the nad1 gene copy numbers of serum and urine samples at all three time points, while for fecal samples the correlations were significant only at day 42 (Fig. 3); this was consistent in both SjP and SjC groups. However, the correlation between infection burdens and the SjR2 gene copy numbers were highly variable and inconsistent amongst the two strain groups (Fig. 3).
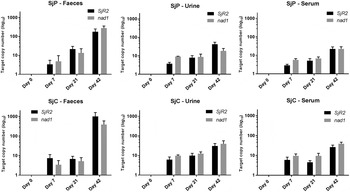
Fig. 2. Variation of target (SjR2 and nad1) S. japonicum DNA copy numbers in mouse serum, urine and feces samples collected at different time points over the course of the infection. Error bars represent the standard error of the mean with n = 6.

Fig. 3. Heat map showing the correlation between DNA quantity and infection burden (worm and egg counts) at different infection time points for the nad1 and SjR2 genes. Each cell indicates the Pearson's correlation coefficient value (r) between the two parameters compared. * Statistically significant correlations (P < 0·05).
Both SjR2 and nad1 were detected in salivary gland DNA from the additional SjP-infected mouse group (Fig. 4). However, the copy numbers of both genes did not show significant correlations with infection burden as determined by egg and worm counts.

Fig. 4. Detection of S. japonicum infection using DNA from the salivary glands of infected mice. (A) Target DNA copy numbers (per 20 μl reaction volume) detected with individual mouse salivary gland samples. (B) Average target DNA copy numbers with error bars representing the standard error of the mean with n = 6.
ddPCR analysis of culture supernatants
The SjR2 and nad1 sequences were consistently detected in SjP schistosomula and adult worm culture supernatants. Target DNA copy numbers of schistosomula culture supernatants collected on day one were relatively higher than those collected on days 2–5. In contrast, adult worm culture supernatant target DNA copy numbers were consistent for all five days (Fig. 5).

Fig. 5. Presence of S. japonicum DNA in (A) adult worm and (B) schistosomula culture supernatants. The DNA copy numbers (per 20 μl reaction volume) for each day correspond to four groups of 100 schistosomula and four groups of five pairs of adult worms. Error bars represent the standard error of the mean with n = 4.
Conventional PCR
Fecal, urine, serum and salivary gland samples from mice and the adult and schistosomula supernatants subjected to cPCR of SjR2 and nad1 showed the expected band sizes of 191 and 82 bp, respectively, for the target amplicons.
DISCUSSION
In this study, we validated the applicability of a novel duplex ddPCR assay we developed (Weerakoon et al. Reference Weerakoon, Gordon, Gobert, Cai and McManus2016) for the diagnosis of S. japonicum infection using a mouse model. The assay is capable of detecting the infection at a very early stage after cercarial penetration of the mammalian host, using conveniently obtainable samples such as urine, saliva and blood. Additionally, the target DNA copy numbers obtained from the assay showed a positive correlation with the infection burden assessed by direct parasitology. The assay proved applicable for both SjC and SjP geographical strain infections.
A higher challenge dose of cercariae was used in the experimental infection of mice with SjP than SjC due to the known higher virulence of the latter (Hope et al. Reference Hope, Duke and McManus1996). Resulting infection burdens in terms of egg and worm pair counts were higher with SjC, consistent with previous findings (Hope et al. Reference Hope, Duke and McManus1996). These and other phenotypical differences between the SjC and SjP geographical strains have been investigated using transcriptomic and comparative genomic methods (Moertel et al. Reference Moertel, McManus, Piva, Young, McInnes and Gobert2006; Gobert et al. Reference Gobert, You, Jones, McInnes and McManus2013).
The mouse model used in this study demonstrated that S. japonicum DNA was detectable in the stool, serum and urine of infected animals from day 7 post-cercarial challenge infection. This is important as the ability to detect a pre-patent S. japonicum infection at a very early stage using conveniently obtainable clinical samples with this novel assay clearly has practical significance. Detection of DNA in feces is usually attributed to the presence of eggs, but here we have shown positive identification of S. japonicum DNA in fecal samples well before eggs would have been expected to be found in the host gastrointestinal tract. Moreover, the presence of schistosome DNA in the salivary glands of infected mice suggests it is likely to be excreted in saliva, thereby complementing the previous, albeit limited attempts to detect schistosome DNA in human saliva (Kato-Hayashi et al. Reference Kato-Hayashi, Yasuda, Yuasa, Isaka, Haruki, Ohmae, Osada, Kanazawa and Chigusa2013). Our detection of parasite DNA in blood and urine, as well as in feces at days 7 and 21, are indicative of the presence of cfDNA in these samples. Fecal samples at day 42 contained eggs and therefore it is likely that PCR amplification was mainly due to egg DNA rather than cfDNA. The presence of parasite eggs in peripheral blood and urine was excluded by microscopy of digested blood clots (after serum was removed for DNA extraction and PCR analysis) and urine sediments (after the supernatant was removed for DNA extraction and PCR analysis) for eggs; were found all to be negative. Detectable S. japonicum DNA in fecal samples collected at days 7 and 21 (egg negative by microscopy) and in all urine, serum and salivary gland samples would likely be due to parasite cfDNA. Such cfDNA could originate from schistosomula, adult worms or eggs. The different stages of S. japonicum within the mammalian host are in direct contact with the circulatory system, into which it is likely that parasite cellular material or fragments are constantly shed, so that S. japonicum DNA will be found in relatively high abundance and can be extracted from blood and other bodily fluids.
Parasite DNA detected in the first three weeks of infection likely originates from adult worms or schistosomula and not eggs. DNA in sera may come from the residual cellular debris of dead schistosomula when they migrate through the circulation, the tegumental shedding of growing worms and/or schistosome excretory/secretory products (Mulvenna et al. Reference Mulvenna, Moertel, Jones, Nawaratna, Lovas, Gobert, Colgrave, Jones, Loukas and McManus2010; Xu et al. Reference Xu, Liu, Guo, Wang, Qiu, Sun, Guan, Zhu, Xia and Wu2013; Weerakoon and McManus, Reference Weerakoon and McManus2016). Previous studies indicate that the amount of schistosome cfDNA in the circulation drops significantly following praziquantel treatment indicating that the major source of cfDNA is from live adult worms (Wichmann et al. Reference Wichmann, Panning, Quack, Kramme, Burchard, Grevelding and Drosten2009; Xu et al. Reference Xu, Liu, Guo, Wang, Qiu, Sun, Guan, Zhu, Xia and Wu2013). These studies further showed that schistosome cfDNA persisted for several months following praziquantel therapy, indicating the possible slow release of cfDNA from inactive eggs (Wichmann et al. Reference Wichmann, Panning, Quack, Kramme, Burchard, Grevelding and Drosten2009; Xu et al. Reference Xu, Liu, Guo, Wang, Qiu, Sun, Guan, Zhu, Xia and Wu2013). Therefore, the use of cfDNA to assess therapeutic success will require testing pre-treatment and post-treatment as slow release of DNA from inactive eggs will show as positive for several months. Comparing pre- and post-treatment gene copy numbers will allow for correct assessment of treatment success by showing a reduction in the gene copy number following treatment.
It is still unclear how DNA from adult schistosome worms arrives in the intestinal tract from the circulation (Oliveira et al. Reference Oliveira, Santos, Gonçalves, Barreto and Peralta2010; Gordon et al. Reference Gordon, Acosta, Gobert, Olveda, Ross, Williams, Gray, Harn, Li and McManus2015a ). One possible explanation could be that schistosomula enter alveolar spaces from pulmonary capillaries and are then moved along the respiratory tract and are swallowed so they, or their DNA, end up in the gastrointestinal tract. However, somules have never been found in the alveolar space. In acute schistosomiasis (Katayama syndrome), a systemic hypersensitivity reaction against migrating schistosomula and early oviposition by adults can cause pathological changes in the gastrointestinal and respiratory tracts in humans, including patchy pulmonary infiltrates and gastrointestinal bleeding, which may result in parasite material ending up in feces (Ross et al. Reference Ross, Bartley, Sleigh, Olds, Li, Williams and McManus2002, Reference Ross, Vickers, Olds, Shah and McManus2007). Intestinal permeability can also be increased with systemic immune hyperactivity (Arrieta et al. Reference Arrieta, Bistritz and Meddings2006). In the early phase of infection, schistosome cfDNA is able to transit from the circulation to the gut lumen, but the precise mechanism, particularly at the pre-oviposition stage requires further exploration.
The presence of parasite DNA in culture supernatants of both adult worms and schistosomula strengthens the argument that live schistosomes release cfDNA as a result of shedding of the tegument, gut excretory secretory products and/or parasite derived exosomes (Twu and Johnson, Reference Twu and Johnson2014; Schorey et al. Reference Schorey, Cheng, Singh and Smith2015; Weerakoon and McManus, Reference Weerakoon and McManus2016). The higher level of schistosomula DNA being released on the first day of cultivation could be due to the likely damage and mortality of parasites following the mechanical transformation of cercariae. The presence of dead or dying schistosomula during culture could not be completely excluded due to the large number of larvae cultivated. However, the subsequent consistent presence of S. japonicum DNA, following the daily renewal of almost all of the culture medium, argues strongly that DNA was released from the parasites into the surrounding environment. This argument gains further support from the results of the cultivated adult worm pairs all of which were alive throughout all 5 days of culture with consistent gene copy numbers being obtained on each day.
The duplex ddPCR assay described here is highly sensitive and can detect very low concentrations of S. japonicum DNA. Importantly, the assay is able to quantify parasite burden using DNA copy numbers. The assay directly calculates the target gene copy numbers, and a new concept in assessing the infection intensity/burden can now be based on DNA copy numbers rather than having to convert the molecular counts back to EPG, as has been done previously with qPCR-based diagnostic assays developed for schistosomes and soil-transmitted helminth infections (Gordon et al. Reference Gordon, Acosta, Gobert, Olveda, Ross, Williams, Gray, Harn, Li and McManus2015a , Reference Gordon, McManus, Acosta, Olveda, Williams, Ross, Gray and Gobert b ; Llewellyn et al. Reference Llewellyn, Inpankaew, Nery, Gray, Verweij, Clements, Gomes, Traub and McCarthy2016). ddPCR exhibits greater precision and sensitivity than qPCR, which is important in the detection of parasite cfDNA as the number of DNA copy numbers in different bodily fluids would be low. The overall cost for ddPCR is slightly higher than qPCR, but the cost for the current duplex ddPCR assay is comparable with qPCR as the chemistry used here uses dsDNA-binding dye (EvaGreen) rather than a fluorescent hydrolysis probe required for multiplexing in qPCR assays. As the newer ddPCR technology matures, its costs are also likely to reduce (Hall Sedlak and Jerome, Reference Hall Sedlak and Jerome2014; Yang et al. Reference Yang, Paparini, Monis and Ryan2014; Huggett et al. Reference Huggett, Cowen and Foy2015).
The current assay exhibited a significant correlation between the infection burden and nad1, but not SjR2, copy numbers. Therefore, the nad1 gene copy number would be an important parameter in determining the burden of infection, which is currently assessed in terms of fecal egg counts (WHO, 2013). Whereas the gene copy numbers equivalent to the thresholds of egg densities (in determining the classes of schistosome intensity) could not be considered within this study, due to the limited range of parasite burden in the infected mice and the small sample size, this could potentially be determined in further assay validation within a human cohort of infected subjects with a higher individual variability of infection intensities. Moreover, taking into account the small body size of a mouse and the relatively high cercarial load used in the infection, the results obtained from the mouse model may not necessarily reflect those of a natural human infection so that validation of the assay using human clinical samples is imperative. Nevertheless, since the assay has a high level of sensitivity and accuracy (Weerakoon et al. Reference Weerakoon, Gordon, Gobert, Cai and McManus2016) we anticipate that very low amounts of parasite cfDNA would be detectable in human clinical samples.
The SjR2 gene showed variable correlation between copy number and infection intensity at different time points and is thus not a good determinant of infection intensity. This inconsistency may be due to the variable nature of retrotransposons within the genome. In particular, the capacity of retrotransposons to proliferate within the genome allows for a higher chances of variable copy numbers (Laha et al. Reference Laha, Brindley, Smout, Verity, McManus and Loukas2002; Schistosoma japonicum Genome Sequencing and Functional Analysis Consortium, 2009; Han, Reference Han2010; Finnegan, Reference Finnegan2012; Coates et al. Reference Coates, Fraser, French and Sappington2013). However, the details on the amounts and the exact mechanisms involved in the regulation of genome retrotransposon copy number is intricate and, as yet, unclear. The ddPCR splits a PCR mixture containing primers, master mix and DNA into 20 000 droplets, within each amplification occurs. In theory, each droplet will contain a single copy of target DNA, which will be amplified, or zero copies of target DNA with no amplification resulting (Hudecova, Reference Hudecova2015; Weerakoon et al. Reference Weerakoon, Gordon, Gobert, Cai and McManus2016). This ddPCR method, therefore, precisely provides the initial number of gene copies in a given sample, as each positive droplet will be counted as one. When retrotransposons are analysed, a DNA fragment within a droplet can contain multiple copies of the retrotransposon genes, but as the amplification occurs within a single droplet all these would be counted as one positive droplet; therefore, the actual gene abundance would not always be accurately represented. Locations of retrotransposons within a DNA strand and positions of fragmentation will therefore determine the reflection of abundance in the ddPCR (Hindson et al. Reference Hindson, Ness, Masquelier, Belgrader, Heredia, Makarewicz, Bright, Lucero, Hiddessen, Legler, Kitano, Hodel, Petersen, Wyatt, Steenblock, Shah, Bousse, Troup, Mellen, Wittmann, Erndt, Cauley, Koehler, So, Dube, Rose, Montesclaros, Wang, Stumbo and Hodges2011). However, SjR2 and other retrotransposons are highly abundant and sensitive makers (Laha et al. Reference Laha, Brindley, Smout, Verity, McManus and Loukas2002; Xia et al. Reference Xia, Rong, Lu, Shi, Xu, Zhang, Gong and Luo2009; Xu et al. Reference Xu, Rong, Zhang, Shi, Zhu and Xia2010; Guo et al. Reference Guo, Zheng, Xu, Zhu, Wang and Xia2012) and cfDNA is generally highly fragmented. The use of highly abundant markers such as retrotransposons increases the likelihood of the target DNA being identified in cfDNA. Overall, while the nad1 gene is more suitable for determining intensity of infection using copy number than SjR2, retrotransposons still represent a valuable resource when determining host infection status.
Concluding comments
Here we have demonstrated a novel ddPCR-based duplex assay, which targets two gene sequences, can be used for the early and accurate diagnosis of schistosomiasis japonica. Furthermore, the potential of the assay to detect parasite DNA in urine and saliva has important practical significance for large-scale epidemiological screening programmes in the future, as collection of these bodily fluids is non-invasive, painless, convenient and cost-effective. Hence, the assay would be a valuable tool for the diagnosis of schistosomiasis japonica, following further validation in a human clinical cohort. Despite its inherent cost, the assay represents a useful complement to current conventional diagnostics for the surveillance of low-intensity S. japonicum infections in lowly prevalent areas, as is now the case in China (Utzinger et al. Reference Utzinger, Zhou, Chen and Bergquist2005; Bergquist and Tanner, Reference Bergquist and Tanner2010), and would be an important tool in monitoring future schistosomiasis japonica outbreaks in controlled areas and in the early diagnosis of pre-patent infections. It is reasonable to speculate that a similar type of duplex assay would be an effective tool in the diagnosis of single and mixed Schistosoma mansoni and Schistosoma haematobium infections in co-endemic areas.
FINANCIAL SUPPORT
The project received funding support from the National Health and Medical Research Council (NHMRC) of Australia (Grant numbers: ID613671; APP1037304; APP1098244). D.P.M. is a NHMRC Senior Principal Research Fellow and Senior Scientist at QIMR Berghofer Medical Research Institute.






