


Small arteries (lumen <300 µm) are responsible for blood pressure control and regional distribution of blood flow, through effects on vascular resistance (Refs Reference Christensen and Mulvany1, Reference Martinez-Lemus, Hill and Meininger2). The lumen of resistance arteries is a function of vasoconstriction exerted by vascular smooth muscle cells (VSMCs) and the structural characteristics of the vessel. Vasomotor control (contraction/relaxation) underlies rapid adaptation of vessel diameter, whereas alterations in structure constitute a dynamic process occurring in response to long-term haemodynamic modifications. Initially structural changes are adaptive but subsequently they become maladaptive, resulting in perturbations in media thickness and lumen diameter (Ref. Reference Gibbons and Dzau3). This process, called vascular remodelling, contributes to the pathophysiology of vascular diseases, including hypertension (Refs Reference Mulvany4, Reference Loirand, Rolli-Derkinderen and Pacaud5, Reference Buus6).
Vascular remodelling is associated with alterations of the luminal diameter (outward or inward) and changes in media mass (hypertrophic, eutrophic or hypotrophic), due to increased transmural pressure and blood flow (Refs Reference Schiffrin7, Reference Virdis8, Reference Baumbach, Sigmund and Faraci9, Reference De Mey10). Whether increased pressure itself or other factors are responsible for the initiation of vascular remodelling remains unclear, but the endothelium probably has an important role because it is a sensor of haemodynamic and humoral factors and is a moderator of signals to underlying VSMCs, critically involved in remodelling (Ref. Reference Xu11). Altered VSMC growth or apoptosis, contraction or relaxation, senescence, calcification, production or degradation of extracellular matrix, and inflammation result in structural changes. Cellular mechanisms and intracellular signalling implicated in remodelling in hypertension are complex and multifactorial, but among the many systems the renin–angiotensin system (RAS) is especially important (Refs Reference Schiffrin and Touyz12, Reference Ferrario13).
The RAS – an update
Classically, the RAS is considered as an endocrine system where angiotensin II (Ang II) is the product of sequential enzymatic cleavage of angiotensinogen. Circulating renin converts hepatic-derived angiotensinogen to angiotensin I (Ang I), which in turn is cleaved by angiotensin-converting enzyme (ACE) to form Ang II. Ang II mediates its biological effects through two G-protein-coupled receptors (GPCRs): Ang II type 1 receptor (AT1R) and Ang II type 2 receptor (AT2R) (Fig. 1). Until recently, Ang II was considered the major effector peptide of the RAS (Ref. Reference Steckelings14). However, the conventional view of the RAS has undergone significant change at three main levels, through: (1) discovery of a functionally active tissue-based RAS; (2) identification of renin/prorenin receptor [(P)RR] and ACE2; and (3) recognition of functionally active Ang-II-derived peptides (Ref. Reference Lacolley15) (Figs 1 and 2).
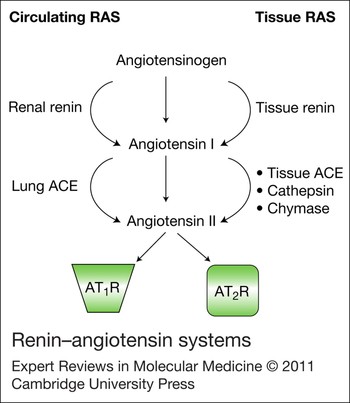
Figure 1. Renin–angiotensin systems. In addition to the classical circulating renin–angiotensin system (RAS), a dynamic tissue RAS has been characterised. Components of the RAS, including enzymes that convert angiotensinogen to angiotensin II (Ang II) through angiotensin I, as well as receptors for Ang II, have been identified in many tissues and organs, including the brain, kidney, eye, heart, vascular system and reproductive organs. Ang II derived from circulating and tissue-based RAS contributes to pathophysiological effects of Ang II. Abbreviations: ACE, angiotensin-converting enzyme; AT1R, Ang II type 1 receptor; AT2R, Ang II type 2 receptor.

Figure 2. Novel components of the renin–angiotensin system and angiotensin receptors. In addition to the classical renin–angiotensin system (RAS; yellow), where angiotensin II (Ang II) is produced through sequential hydrolysis of angiotensinogen by renin and angiotensin-converting enzyme (ACE), Ang II can be produced through non-ACE-dependent systems, and Ang I and II can be metabolised to biologically active small peptides (blue) by ACE2 or other peptidases, as shown. Ang-(1-7) binds to the receptor MAS. Furthermore, Ang II can be metabolised by AMP to Ang III, which, like Ang II, binds to the receptors AT1R and AT2R; and the action of AMPs on Ang II and III can also generate Ang IV, which acts through AT1R, and also by binding to IRAP (although the mechanisms involved remain to be clarified). Renin also acts as a RAS agonist by binding to the renin/prorenin receptor [(P)RR]. Abbreviations: AMP, aminopeptidase; IRAP, insulin-regulated aminopeptidase; NEP, neutral-endopeptidase 24.11; PCP, prolylcarboxypeptidase; PEP, prolylendopeptidase.
Tissue-based RAS
The RAS was originally described as a haemodynamic regulator that increases blood pressure acutely by vasoconstriction and chronically by aldosterone-mediated volume expansion (Ref. Reference Lacolley15). It is now evident that a tissue-based RAS exists that is independently controlled from the circulation RAS (Ref. Reference Bader16). A tissue system is characterised by the presence of all RAS components, including angiotensinogen, renin, ACE, Ang I, Ang II and Ang II receptors (Fig. 1), and is found in the heart, vessels, kidney, adrenal gland, pancreas, central nervous system, reproductive system, lymphatic system and adipose tissue (Refs Reference Thatcher17, Reference Cuadra18). Components of the RAS have also been identified in the eye, which may be important physiologically in maintaining ocular pressure (Ref. Reference Vaajanen19) and pathologically in vasculopathies associated with retinopathy of hypertension and diabetes (Refs Reference Sharma20, Reference Fletcher21), The local RAS acts in an autocrine/intracrine, paracrine and endocrine manner. Elevated tissue levels of RAS components occur in cardiovascular disease independently of blood pressure elevation, such as atherosclerosis, myocardial infarction, cardiac failure, diabetes and kidney disease (Ref. Reference Ribeiro-Oliveira22). There has also been much interest in an intracellular RAS that might contribute to cardiovascular function (Ref. Reference Kumar, Singh and Baker23). Overexpression of intracellular Ang II, using a novel fluorescent fusion protein of Ang II, correlates with elevated blood pressure and kidney pathology (Ref. Reference Redding24).
New RAS players: (P)RR and ACE2
Renin, a protease enzyme produced by the juxtaglomerular apparatus in the kidney, acts on angiotensinogen to form Ang I. It is the key enzyme of the RAS because of its rate-limiting hydrolytic activity (Ref. Reference Castrop25). Renin also acts as a RAS agonist by binding to the (P)RR (Ref. Reference Nguyen and Muller26). (P)RR is a new RAS member that binds renin and prorenin. Prorenin bound to (P)RR undergoes a conformational change and becomes catalytically active to induce activation of signalling pathways, such as mitogen-activated protein kinase (MAPK) pathways, which promote cell growth and fibrosis independently of Ang II in cardiomyocytes, mesangial cells, podocytes, distal tubular cells, vascular endothelial cells and VSMCs (Refs Reference Danser and Nguyen27, Reference Reudelhuber28). The exact (patho)physiological significance of this receptor remains unclear, but it might be important in early development, especially neuronal development. Ang-II-independent (P)RR signalling exacerbates neurotoxin-induced dopaminergic cell loss, suggesting a role for (P)RR in neural pathologies such as Parkinson disease (Ref. Reference Valenzuela29). Transgenic animals overexpressing (P)RR develop high blood pressure or glomerulosclerosis, and increased expression of (P)RR occurs in models of hypertension (Ref. Reference Danser and Nguyen27). However, definitive proof of a role for this receptor in cardiovascular disease is still lacking, with studies failing to demonstrate a significant function of (P)RR in hypertension and target organ damage (Ref. Reference Reudelhuber28).
(P)RR is a multifunctional receptor that mediates effects beyond the RAS (Ref. Reference Hitom, Liu and Nishiyama30). A truncated form of the receptor, termed M8.9, might be an accessory protein of vacuolar H+-ATPase (V-ATPase) involved in non-RAS-related functions (Ref. Reference Ludwig31).(P)RR/ATP6AP2 is essential for V-ATPase assembly in cardiomyocytes (Refs Reference Kinouchi32, Reference Advani33, Reference Nguyen34). V-ATPase has a role in physiological and biochemical processes by regulating intracellular pH, an effect that is enhanced by (pro)renin binding to (P)RR (Ref. Reference Cruciat35). (P)RR also functions in a (pro)renin-independent manner as an adaptor protein between V-ATPase and WNT receptors in WNT signalling (Ref. Reference Cruciat35). WNT signalling is involved in cell proliferation, polarity and fate determination during embryonic development and tissue homeostasis and has been implicated in various pathologies, including cancer.
ACE is the major Ang-II-generating enzyme. It also inactivates kinins, thereby reducing bradykinin-mediated vasodilation. ACE2 catalyses Ang I and Ang II to generate the Ang peptides Ang-(1–9) and Ang-(1-7), which mediate, in general, effects opposite to those of Ang II (Refs Reference Cruciat35, Reference Imai36). Reduced ACE2, and consequent decreased Ang-(1-7) production, is associated with vasoconstriction, vascular remodelling and oxidative injury in hypertension, diabetes and kidney disease (Refs Reference Imai36, Reference Dilauro37). By contrast, increased ACE2 activity promotes vasodilation and as such is currently being tested as a potential therapeutic target (Ref. Reference Calò38).
Ang-II-derived small peptides: Ang-(1-7), Ang III, Ang IV and other Ang peptides
Ang-(1-7) is formed from Ang II by prolylendopeptidase, prolylcarboxypeptidase or ACE2, or directly from Ang I through hydrolysis by prolylendopeptidase and endopeptidase 24.11 (Ref. Reference Cangussu39) (Fig. 2). It is metabolised by ACE to Ang-(1-5). Ang-(1-7) is present in the circulation and in tissues (brain, heart, kidneys, vessels, liver, reproductive organs) (Refs Reference Pereira40, Reference Reis41), and binds to its GPCR MAS (Ref. Reference Yang51). In general, Ang-(1-7) opposes Ang II actions, by mediating vasodilation, growth inhibition, anti-inflammatory responses, and antiarrhythmogenic and antithrombotic effects (Refs Reference Santiago42, Reference Sampaio43), through nitric oxide synthase (NOS)-derived NO production, activation of protein tyrosine phosphatases (PTPs), inhibition of MAPKs and inhibition of NADPH oxidase (Refs Reference Sampaio43, Reference Gava44). Ang-(1-7)–MAS can hetero-oligomerise with AT1R, thereby inhibiting Ang II actions. The ACE2–Ang-(1-7)–MAS axis is now considered as a counter-regulatory system to the ACE–Ang-II–AT1R axis. There has been enormous development in the field of Ang-(1-7) biology, as reviewed in Refs Reference Santos, Ferreira and Simões45 and Reference Santos, Frézard and Ferreira46.
Ang III, formed from Ang II by aminopeptidase A, is a biologically active peptide with actions similar to those of Ang II (Ref. Reference Reaux, Fournie-Zaluski and Llorens-Cortes47). Ang III infusion in experimental models and in humans increases blood pressure, promotes vasoconstriction and stimulates aldosterone production (Ref. Reference Healy and Song48). In vitro, it stimulates growth, production of pro-inflammatory mediators and deposition of extracellular matrix proteins (Ref. Reference Stragier49). Ang III binds to AT1R and to AT2R.
Another small peptide derived from Ang II is Ang IV, generated by aminopeptidase N, which removes arginine from the N-terminus of Ang III (Ref. Reference Wright and Harding50). Ang IV can also be formed directly from Ang II by the enzyme d-aminopeptidase. Ang IV increases renal blood flow, induces vasodilation and improves cardiac function (Ref. Reference Yang51). Ang IV also exerts a wide range of neural effects, including enhancement of learning and memory recall, and protection against cerebral ischaemia, and it has anticonvulsant and antiepileptogenic properties. Some of these actions are mediated through AT1R, but others are due to binding of Ang IV to a specific binding site, AT4R, which has been identified as the transmembrane enzyme insulin-regulated aminopeptidase (IRAP) (Ref. Reference Demaegdt52). Exact mechanisms of action whereby Ang IV induces effects through IRAP are unclear but might involve Ang IV (1) acting as an inhibitor of the catalytic activity of IRAP, resulting in accumulation of IRAP's neuropeptide substrates, (2) interacting with the uptake of glucose, because IRAP colocalises with the glucose transporter GLUT4, and (3) acting as an agonist through IRAP in a receptor-mediated manner. The biological significance of Ang IV–IRAP remains to be elucidated.
Other angiotensin peptides include Ang-(3-7), important in the brain and kidney (Ref. Reference Ferreira, Souza Dos Santos and Campagnole-Santos53), Ang-(1-9), which enhances bradykinin actions, NO production and platelet regulation (Ref. Reference Chappell54), and Ang-(1-12), implicated in cardiac function (Ref. Reference Ferrario55).
Angiotensin receptors
Ang II binds with high affinity to two distinct receptor subtypes – AT1R and AT2R (Fig. 3) – which are members of the seven-transmembrane-domain GPCR superfamily. AT1R has high affinity for biphenylimidazoles (e.g. losartan) and AT2R has high affinity for tetrahydroimidazopyridines (e.g. PD123177 and its derivative CGP42112).

Figure 3. Molecular and cellular processes mediated by angiotensin II and Ang-(1-7) that influence vascular changes in hypertension. Angiotensin II (Ang II) binds with high affinity to its two distinct receptor subtypes, AT1R and AT2R, whereas Ang-(1-7) binds to the receptor MAS. Effects mediated by Ang II through AT1R are, in general, opposed by signalling through AT2R and MAS. AT1R and AT2R are negatively regulated by G-protein-coupled-receptor-interacting proteins (GIPs), including: AT1R-associated protein (ATRAP) and AT1R-associated protein 1 (ARAP1), which interact with AT1R; and the AT2R-interacting protein (ATIP), which interacts with AT2R.
AT1R interacts with multiple heterotrimeric G proteins, including Gq/11, Gi, G12 and G13, and produces second messengers such as inositol trisphosphate, diacylglycerol and reactive oxygen species (ROS) (Ref. Reference Higuchi56). It also activates receptor and nonreceptor tyrosine kinases, serine/threonine kinases, MAPKs, p70S6K (RPS6KB2), AKT (protein kinase B) and protein kinase C (PKC) (Ref. Reference Higuchi56). AT1R mediates most of the pathophysiological effects of Ang II, including vasoconstriction, inflammation, growth and fibrosis, whereas AT2R counteracts many AT1R-mediated actions (Ref. Reference Steckelings, Kaschina and Unger57).
Ang II type 1 receptors
In humans, the gene encoding AT1R (AGTR1) is located on chromosome 3, whereas in rodents, two genes on chromosomes 17 and 2 encode two subtypes: AT1AR and AT1BR. AT1R is expressed primarily in VSMCs, liver, kidney, heart, lung, adrenal cortex, pituitary, brain and adipose tissue (Refs Reference Steckelings, Kaschina and Unger57, Reference deGasparo58, Reference Cassis59). Although the function of Ang-II–AT1R in adipose tissue is still unclear, it has been implicated in adipocyte maturation and in adipocytokine secretion (Refs Reference Steckelings, Kaschina and Unger57, Reference deGasparo58, Reference Cassis59). Ang II binding to AT1R results in coupling of G proteins (Gq/G11 or Gi/Go) to the C-terminus and to stimulation of signalling pathways, including phospholipases C, D and A2 (PLC, PLD and PLA2), Ca2+ channels, adenylate cyclase, MAPKs, JAK–STAT (Janus kinase and ‘signal transducers and activators of transcription’) and NADPH oxidase (Refs Reference Higuchi56, Reference Yiannikouris60).
GPCRs interact not only with G proteins but also with accessory proteins, termed GPCR-interacting proteins (GIPs). A GIP specifically associated with AT1R, called AT1R-associated protein (ATRAP), acts as a negative regulator of AT1R (Ref. Reference Min61) (Fig. 3). ATRAP is expressed in the aorta, heart, lung and kidney. Overexpression leads to decreased cell proliferation. ATRAP-transgenic mice show reduced neointimal formation, and decreased inflammation and oxidative stress in response to Ang II, supporting the inhibitory role of ATRAP in cardiovascular remodelling (Ref. Reference Anaka62). Moreover, in ATRAP−/− mice, blood pressure and plasma volume are increased (Ref. Reference Oppermann63). Another identified GIP, ARAP1 (AT1R-associated protein 1), is involved in the increase in membrane AT1R number following Ang II stimulation and in the recycling of the receptor (Refs Reference Mogi, Iwai and Horiuchi64, Reference Tanaka65). For recent reviews of the AT1R, see Refs Reference Higuchi56 and Reference Mogi, Iwai and Horiuchi66.
Ang II type 2 receptors
The gene encoding AT2R (AGTR2) is located on the X chromosome. AT2R shares partial amino acid homology (34%) with AT1R (Ref. Reference Grady67). AT2R is the predominant Ang II receptor in the fetus (Ref. Reference Grady67). After birth, this changes and AT1R becomes the dominant subtype (Ref. Reference Horiuchi, Akishita and Dzau68). AT2R expression is low in a few organs postnatally, including the adrenal gland, ovary, brain, kidney, cardiomyocytes and peripheral vasculature (Refs Reference Savoia69, Reference Hu70). Increased AT2R expression occurs in pathological conditions, including hypertension, myocardial infarction, cardiac failure, renal failure, cerebral ischaemia and diabetes (Refs Reference Ohkubo71, Reference Bautista72). Metabolic factors, hormones and cytokines may upregulate AT2R expression, whereas glucocorticoids and growth factors reduce AT2R expression. AT1R stimulation interferes with AT2R expression, suggesting crosstalk between the two receptors (Refs Reference Miura and Karnik73, Reference De Paolis74). In vivo studies show that AT1R blockade is associated with AT2R upregulation, which may mediate effects of increased unopposed Ang II.
Few studies have investigated human AT2R biology. In healthy human skin, AT2R is expressed throughout the epidermal and dermal structures (Ref. Reference Steckelings75), and is upregulated in wounds (Ref. Reference Steckelings76). In the lung, AT2R is present in epithelial cells, mucous glands, vascular endothelial cells, fibroblasts and macrophages (Ref. Reference Bullock77). In the kidney, AT2R is expressed in interlobular arteries (Ref. Reference Matsubara78). Although there is no direct evidence in humans, it has been proposed that AT2R inhibits renin biosynthesis and Ang II formation (Ref. Reference Carey79) and regulates glomerular blood flow and pressure natriuresis (Ref. Reference Carey and Siragy80). In the heart, AT2Rs are present in the myocardium (Ref. Reference Tsutsumi81) and in the coronary vascular bed of subjects with no evidence of cardiovascular disease (Ref. Reference Wharton82), where they mediate vasodilation (Ref. Reference Batenburg83). AT2R is increased (Ref. Reference Tsutsumi81), decreased (Ref. Reference Regitz-Zagrosek84) or unchanged (Ref. Reference Haywood85) in patients with cardiac failure. Because AT1Rs decrease with cardiac dysfunction (Refs Reference Steckelings76, Reference Tsutsumi81, Reference Wharton82, Reference Batenburg83, Reference Regitz-Zagrosek84, Reference Haywood85), the ratio of AT2R to AT1R tends to be increased in cardiac disease. AT2Rs are expressed in the vascular wall of small resistance-size arteries from hypertensive diabetic patients when patients are treated with Ang II receptor blockers (ARBs) (Ref. Reference Savoia86).
The GIP AT2R-interacting protein (ATIP) is associated with AT2R, and involved in transinactivation of receptor tyrosine kinases and growth inhibition (Ref. Reference Tsutsumi81). ATIP is also known as mitochondrial tumour suppressor gene 1 (MTUS1) and AT2R-binding protein of 50 kDa (ATBP50). ATIP is expressed in tissues in which AT2R is abundant, including the uterus and adrenal tissue (Refs Reference Reinemund87, Reference Di Benedetto88).
Ang II signalling in the cardiovascular system
Ang II mediates effects through complex signalling pathways that are stimulated following binding to its cell-surface receptors (Ref. Reference Touyz and Schiffrin89). Both receptors regulate VSMC function, although they differ in their actions (Fig. 3). Whereas AT1R is associated with growth, inflammation and vasoconstriction, AT2R is associated with apoptosis and vasodilation (Refs Reference Touyz and Schiffrin89, Reference Henrion, Kubis and Lévy90). In pathological conditions, AT2R may also stimulate hypertrophy and inflammation (Ref. Reference Tian91).
Signalling through AT1R
Growth signalling by Ang II in vascular cells
Ang II stimulates cell growth through phosphorylation of MAPKs [ERK1/2 (MAPK3/1), p38MAPK (MAPK14), JNK (MAPK8), ERK5 (MAPK7)] and tyrosine kinases [SRC, JAK, FAK (PTK2), PYK2 (PTK2B), p130Cas (BCAR1)], mobilisation of intracellular Ca2+ and ROS production (Ref. Reference Touyz92). SRC is an important early signalling molecule involved in trophic and contractile actions of Ang II in hypertension (Refs Reference Touyz, Yao and Schiffrin93, Reference Touyz94). In cardiac, renal and vascular tissue from hypertensive rats and VSMCs from hypertensive patients, Ang-II-stimulated activation of tyrosine kinases and MAPKs is augmented (Refs Reference Touyz95, Reference Frank96), contributing to remodelling through enhanced growth, inflammation, fibrosis and constriction.
Ang II also activates receptor tyrosine kinases, even though it may not bind directly to these receptors. This process of transactivation has been demonstrated for epidermal growth factor (EGF) receptor, platelet-derived growth factor (PDGF) receptor and insulin-like growth factor 1 (IGF1) receptor (Refs Reference Saito and Berk97, Reference Touyz98). Ang-II-induced transactivation occurs through tyrosine kinases (PYK2 and SRC), redox-sensitive processes and a metalloprotease-dependent shedding of heparin-binding EGF-like growth factor (HB-EGF) (Refs Reference Montezano99, Reference Mifune100). The metalloprotease responsible for this process is ADAM17 (Refs Reference Ohtsu101, Reference Melenhorst102). Ang II also increases the production of vasoactive hormones and growth factors in hypertension, such as endothelin 1 (ET-1/EDN1), PDGF, transforming growth factor β (TGF-β), basic fibroblast growth factor (bFGF/FGF2) and IGF1, which could promote hyperplasia and fibrosis, further contributing to growth in arterial remodelling.
Ang II signalling through RhoA/Rhokinase
Activation of RhoA (RHOA) and its downstream target Rhokinase (ROCK) is an important mechanism of vasoconstriction by Ang II and has been implicated in the pathophysiology of hypertension and other vascular diseases (Refs Reference Loirand, Guerin and Pacaud103, Reference Loirand and Pacaud104). RhoA, a member of the Rho family of small GTPase proteins, is abundantly expressed in VSMCs and participates in vasoconstriction through phosphorylation of myosin light chain and sensitisation of contractile proteins to Ca2+. In VSMCs, Ang-II–AT1R increases RhoA activity (Refs Reference Uehata105, Reference Seko106) through the G12/13 family of G proteins and Gq (Ref. Reference Gohla, Schultz and Offermanns107). RhoGEFs (guanine-nucleotide-exchange factors), such as ARHGEF1 (p115RhoGEF), ARHGEF12 (LARG) and ARHGEF11 (PDZ-RhoGEF), catalyse exchange of GDP for GTP on RhoA and are sensitive to G12/13; hence they have an important role in RhoA activation (Ref. Reference Guilluy108). Ang-II–AT1R induces phosphorylation of ARHGEF1 by JAK2 (Ref. Reference Wirth109). In vivo, deletion of VSMC ARGHEF1 does not modify blood pressure in transgenic mice; however, Ang-II-induced contraction in aortic rings is inhibited (Refs Reference Bregeon110, Reference Mori111).
Inactivation of RhoGAP (GTPase-activating protein; p190A/ARHGAPs) is also important in RhoA/Rhokinase signalling by GPCRs, as shown by the recent identification, in VSMCs, of the tyrosine phosphatase SHP2 (PTPN11) as a novel negative regulator of RhoGAP (Ref. Reference Bregeon110). Activated SHP2 maintains basal p190A activation and consequently a low RhoA/Rhokinase activity in rat aortic SMCs (Ref. Reference Mori111). SHP2 regulation by Ang II through AT1R occurs in a redox-sensitive manner (Refs Reference Touyz112, Reference Tabet113). Under certain conditions, possibly when AT2R is upregulated, Ang II can inhibit RhoA activity to induce vasodilation. This involves AT2R-mediated STE20-like kinase (SLK)-induced phosphorylation of Ser188 of RhoA (Ref. Reference Guilluy114).
Ang-II-induced hypertension in rodents shows increased vascular RhoA/Rhokinase activation, without changes in expression (Ref. Reference Loirand and Pacaud104). This is associated with increased activity of ARHGEF1, thought to be important in RhoA hyperactivation, vasoconstriction and hypertension (Ref. Reference Uehata105). Pharmacological inhibition of Rhokinase with fasudil or Y27632 suppresses acute pressor responses of Ang II, but does not reduce blood pressure chronically, supporting the role of RhoA/Rhokinase in acute vasoconstriction rather than in mechanisms associated with adaptive vascular remodelling in hypertension that occur chronically with Ang II infusion (Refs Reference Loirand, Guerin and Pacaud103, Reference Loirand and Pacaud104).
Ang II signalling through ROS
Ang II mediates many actions by stimulating the formation of ROS – highly reactive, bioactive, short-lived molecules derived from reduction of O2 (Ref. Reference Taniyama and Griendling115) (Fig. 4). Major ROS produced within vascular cells include the superoxide anion (•O2−), H2O2, the peroxide anion (•OH), NO and the peroxynitrite anion (ONOO−) (Refs Reference Guzik, Korbut and Adamek-Guzik116, Reference Harrison117). Among the many ROS generators, including mitochondria, cyclooxygenase, lipoxygenase, haem oxygenase, cytochrome P450 monooxygenase and xanthine oxidase, NAD(P)H (nicotinamide adenine dinucleotide phosphate, reduced form) oxidases are of major importance in vascular cells (Ref. Reference Griendling118). NAD(P)H oxidase is a multisubunit enzyme (Refs Reference Lassegue and Clempus119, Reference Touyz120) that catalyses the production of superoxide by the one-electron reduction of oxygen using NAD(P)H as the electron donor: 2O2 + NAD(P)H → 2•O2− + NAD(P)H + H+. The prototypical NAD(P)H oxidase is found in phagocytes, and is made up of five components: p47phox, p67phox, p40phox, p22phox and gp91phox (phox for ‘phagocyte oxidase’; also known as NCF1, NCF2, NCF4, CYBA and CYBB/NOX2, respectively) (Ref. Reference Babior, Lambeth and Nauseef121). Unlike phagocytic NAD(P)H oxidase, which is activated only on stimulation and which generates superoxide in a burst-like manner extracellularly, vascular oxidases are constitutively active, produce superoxide intracellularly in a slow and sustained manner, and act as intracellular signalling molecules (Refs Reference Lassegue and Clempus119, Reference Touyz120, Reference Babior, Lambeth and Nauseef121). All the phagocytic NAD(P)H oxidase subunits are expressed, to varying degrees, in vascular cells (Refs Reference Rey and Pagano122, Reference Heidari, Shah and Gove123). The newly discovered gp91phox isoforms NOX1, NOX4 and NOX5 [NOX for ‘NAD(P)H oxidase’] have also been detected in the vasculature (Refs Reference Lambeth124, Reference Hilenski125, Reference Maturana, Krause and Demaurex126). NOX1 is implicated in cell growth, NOX4 in delayed cell growth, and gp91phox may act as a proton pump in addition to being part of the NAD(P)H oxidase complex (Refs Reference Hilenski125, Reference Maturana, Krause and Demaurex126). NOX5 is implicated in atherosclerosis (Refs Reference Montezano127, Reference Fulton128). All vascular NOX isoforms, including NOX1, NOX2/gp91phox, NOX4 and NOX5, are regulated by Ang II. The functional significance of different NOXs awaits clarification, but their differential tissue distribution, cellular localisation and subcellular compartmentalisation probably have a major role in NOX-specific actions (Refs Reference Montezano129, Reference Montezano130, Reference Haurani131).

Figure 4. Angiotensin-II-mediated signalling events in vascular smooth muscle cells that promote vascular remodelling and inflammation. Binding of angiotensin II (Ang II) to the cell-surface Ang II receptor AT1R induces activation of NADPH oxidase (a multicomplex oxidase comprising NOX, p22phox, p47phox and p67phox subunits and RAC), which results in increased generation of reactive oxygen species (ROS), particularly superoxide (•O2−) and hydrogen peroxide (H2O2). NADPH oxidase is regulated by SRC tyrosine kinase, protein kinase C (PKC), phospholipase D (PLD) and phospholipase A2 (PLA2). ROS act as second messengers to regulate redox-sensitive signalling molecules, important in inflammation, cell growth and fibrosis. Ang-II–AT1R also induces activation of mitogen-activated protein kinases (MAPKs) and increases intracellular Ca2+, processes that influence contraction, growth, inflammation and fibrosis. The vascular remodelling and inflammation triggered by these Ang-II-mediated signalling events contribute to vascular injury in hypertension.
Ang II is a potent stimulator of vascular NOX (Refs Reference Montezano130, Reference Haurani131, Reference Dikalova132, Reference Pendergrass133). Mechanisms linking Ang II to the enzyme have not been fully elucidated, but PLD, PKC, SRC, phosphoinositide 3-kinase and RAC (AKT1) are important (Refs Reference Touyz92, Reference Rey and Pagano122, Reference Heidari, Shah and Gove123, Reference Djordjevic134). In hypertension, these processes are augmented, contributing to increased NOX activation and oxidative stress (Refs Reference Briones and Touyz135, Reference Cheng136). Polymorphisms in the promoter of the p22phox gene have been identified in hypertensive patients, which may also have a role in increased NAD(P)H-oxidase-driven generation of superoxide in hypertension (Ref. Reference San Jose137).
Superoxide and H2O2 activate multiple signalling molecules, including MAPKs, tyrosine kinases, PTPs and transcription factors (NF-κB, AP-1 and HIF-1) (Refs Reference Nathan138, Reference Torres and Forman139), which regulate cell growth, migration, inflammation, fibrosis and contraction/dilation, important in remodelling in hypertension. ROS modify signalling molecules in large part through oxidative modification of redox-sensitive proteins, such as PTPs (Refs Reference Chiarugi and Cirri140, Reference Knock and Ward141).
Inhibition of NAD(P)H oxidase activity is now considered, at least experimentally, as a putative therapeutic strategy in the treatment of hypertension (Refs Reference Hamilton142, Reference Elmarakby, Williams and Pollock143). In fact, it has been suggested that some of the beneficial actions of ACE inhibitors and AT1R antagonists might be mediated, in part, by decreasing vascular oxidative stress. These effects have been attributed to direct inhibition of NAD(P)H oxidase by ARBs (Ref. Reference Dohi144).
Signalling through AT2R
AT2R stimulation activates the NO–cGMP-dependent pathway (Ref. Reference Abadir, Carey and Siragy145). This occurs either directly or indirectly through bradykinin or increased endothelial NOS activity or expression. AT2R activation is associated with phosphorylation of JNK, PTPs, IκBα (inhibitor of NF-κB) and the transcription factor ATF2, and dephosphorylation of p38MAPK, ERK1/2 and STAT3, which are linked to antiproliferative and anti-inflammatory effects and apoptosis (Refs Reference Savoia146, Reference Hu147, Reference Dandapat148, Reference Brassard, Amiri and Schiffrin149) AT2R may induce relaxation by opening large-conductance Ca2+-activated K+ channels (BKCa) (Ref. Reference Dimitropoulou150) and by negatively regulating vascular Rho/Rhokinase. AT2R also enhances the activity of tyrosine phosphatases and vanadate-sensitive phosphatases MKP1 (DUSP1), SHP1 (PTPN6) and PP2A (Refs Reference Horiuchi151, Reference Bedecs152). In vivo, pharmacological stimulation of AT2R by a novel nonpeptide agonist, compound 21, evokes vasodepressor effects in spontaneously hypertensive rats (Ref. Reference Bosnyak153).
Ang II and microparticles
Microparticles are tiny fragments of cellular membranes that are generated from activated or apoptotic cells (Ref. Reference Tao154). Microparticles derive from various cell types, including platelets, endothelial cells, leukocytes and erythrocytes (Ref. Reference Horstman155), and their circulating levels are increased in many pathological conditions, including hypertension (Refs Reference Nomura156, Reference Preston157). Microparticles may be significant cellular effectors that contribute to disease progression in hypertension, because they can impair vasorelaxation (Refs Reference Brodsky158, Reference Tesse and Martinez159).
Emerging evidence indicates that Ang II may have a role in microparticle formation. AT1R blockade with losartan or valsartan is associated with reduced plasma levels of monocyte-, platelet- and endothelial-cell-derived microparticles in hypertensive patients (Refs Reference Nomura160, Reference Nomura161). Thus, AT1R blockade may interfere with microparticle production. However, whether Ang-II–AT1R can directly stimulate microparticle generation is unclear. In cultured human umbilical vein endothelial cells, Ang II did not stimulate microparticle formation (Refs Reference Labios162, Reference Simak163). Although it is possible that Ang II may directly influence microparticle biology in other cell types, the in vivo reductions by AT1R blockade may be an indirect result of reduced blood pressure rather than a direct, receptor-mediated reduction of microparticle formation. The exact role of Ang II in microparticle biology awaits further clarification.
Ang II and endothelial progenitor cells
Circulating endothelial progenitor cells (EPCs) are bone-marrow-derived cells capable of developing into mature endothelial cells (Ref. Reference Urbich and Dimmeler164). The number of circulating EPCs is an important determinant of endothelial function because decreased numbers are associated with reduced arterial elasticity (Ref. Reference Hill165) and decreased endothelial integrity (Ref. Reference Yu166). Circulating EPCs are reduced in hypertensive and diabetic patients and in hypertensive rats (Refs Reference Yu166, Reference Sen167, Reference Yoshida168). Ang II appears to have paradoxical effects on EPCs (Ref. Reference Qian169). On the one hand, acute Ang II administration synergistically increases vascular endothelial growth factor (VEGF)-mediated proliferation of EPCs in an AT1R-dependent manner (Ref. Reference Imanishi170). In Ang-II-infused mice, the number of circulating EPCs increases, an effect blocked by AT1R antagonism, possibly through decreased ROS formation (Ref. Reference Salguero171). Ang II and VEGF synergistically increase EPC proliferation, possibly due to inhibition of EPC apoptosis (Ref. Reference Yin172).
On the other hand, there is also evidence that Ang II decreases EPC number. Olmesartan increased EPC number in patients with diabetes (Ref. Reference Urbich and Dimmeler164), whereas Ang II infusion in rats decreased EPC number, which was reversible by valsartan (Ref. Reference Kobayashi173). Interestingly, Ang II infusion in these rats reduced telomerase activity and accelerated cellular senescence in EPCs, suggesting that reductions in EPCs after Ang II infusion were due to impaired proliferation. Studies in cultured EPCs suggest that ROS are involved in Ang-II-mediated EPC senescence (Refs Reference Imanishi174, Reference Sanada175).
Ang-(1-7) may also have a role in EPC formation. Ang-(1-7) increases EPC number within a population of cultured bone marrow mononuclear cells (Ref. Reference Iusuf176). However, the mechanisms underlying Ang-(1-7) EPC effects are unclear.
Role of Ang II in arterial remodelling
VSMCs are critically involved in maintaining vascular integrity and tone. They are dynamic, plastic, multifunctional cells, which contribute to arterial remodelling through various processes, including growth/apoptosis, reorganisation, altered production of extracellular matrix and inflammation (Ref. Reference Touyz and Schiffrin177). In hypertension, Ang-II-stimulated hyperplasia and hypertrophy contribute to vascular remodelling (Refs Reference Touyz and Schiffrin177, Reference Schiffrin and Touyz178, Reference Touyz179).
Apoptosis and fibrosis also contribute to structural remodelling (Refs Reference Lemay, Hale and deBlois180, Reference Intengan and Schiffrin181). Apoptosis influences fine-tuning of media growth, and is increased in some vascular beds and decreased in others (Ref. Reference Intengan and Schiffrin181). Whether apoptosis is a growth-associated compensatory and adaptive process or a primary event is unclear. However, an imbalance between growth and apoptosis could be important. Detachment of VSMCs and endothelial cells (anoikis), increased microparticles and decreased EPCs might further contribute to vascular dysfunction and remodelling in hypertension (Refs Reference Lemay, Hale and deBlois180, Reference Intengan and Schiffrin181, Reference Diep, Li and Schiffrin182).
Vascular fibrosis involves accumulation of extracellular matrix proteins, such as collagen, elastin, fibronectin and proteoglycans, in the media. Collagen deposition is increased in resistance arteries from hypertensive rats and in patients with hypertension (Ref. Reference Tayebjee, MacFadyen and Lip183). Increased collagen I and III mRNA and enhanced collagen protein synthesis by fibroblasts have also been demonstrated in hypertensive patients (Ref. Reference Delva184). Experimental studies indicate that collagen accumulation is related to increased synthesis (Ref. Reference Odenbach185), decreased degradation by matrix metalloproteinases (MMPs) (Ref. Reference Onal186) and enhanced tissue inhibitor of metalloproteinase 1 (TIMP1) production (Ref. Reference Castro187), processes influenced by Ang II.
Ang-II-elicited growth and profibrotic effects are modulated, in part, by endogenous production of mitogenic factors, such as TGF-β, PDGF, EGF, IGF1 and ET-1 (Refs Reference Sarkar188, Reference Satoh189). Of these, TGF-β, a multifunctional cytokine, appears to be especially important. TGF-β increases extracellular matrix biosynthesis, downregulates matrix degradative enzymes and influences integrin receptors (Ref. Reference Popovic190). TGF-β, synthesised by macrophages, lymphocytes, fibroblasts, VSMCs and renal cells, elicits effects by interacting with two cell-surface membrane signalling receptors: a type II TGF-β receptor (TGFBR2) and a type I receptor (TGFBR1/ALK-5). TGF-β is negatively regulated by unique proto-oncoproteins, SKI and SnoN (SKIL) (Ref. Reference Solomon191). Signals from the activated TGF-β receptor complex are transduced to the nucleus by SMAD proteins (Ref. Reference Luo192). Major downstream profibrogenic mediators of TGF-β include p38MAPK. TGF-β is overexpressed in many cardiovascular and renal disorders associated with activation of the RAS. In support of this, inhibition of the RAS with ACE inhibitors or AT1R blockers correlates with suppression of TGF-β production and amelioration of fibrosis (Ref. Reference Liu and Feng193).
Ang II, inflammation and arterial remodelling
Inflammation participates in vascular remodelling (Refs Reference Viel194, Reference Briones and Touyz195, Reference Jamaluddin196, Reference Tieu197) and might contribute to accelerated vascular damage in cardiovascular diseases and in ageing. Whether Ang II or blood pressure elevation itself, through effects on adhesion molecules, chemokines and cytokines induced by cyclic mechanical strain, is associated with the inflammatory response in hypertension is unclear (Ref. Reference Wung198). Activators of nuclear receptors such as PPARs (peroxisome proliferator-activated receptors), which are hypolipidaemic agents (fibrates, PPARα agonists) or insulin sensitisers (glitazones, PPARγ agonists), downregulate the vascular inflammatory response in experimental animals and decrease serum markers of inflammation in humans (Refs Reference Sigmund199, Reference Sugawara200). Thus, PPARs and vasoactive substances may be endogenous modulators of the inflammatory process involved in vascular structural changes occurring in hypertension. Ang II downregulates PPARs through activation of NF-κB (Ref. Reference Tham201).
Vascular inflammation is characterised by recruitment of monocytes and lymphocytes into the subendothelial space, production of chemotactic cytokines, increased expression of adhesion molecules, reactive VSMC proliferation, decreased EPCs, and altered extracellular matrix production and degradation (Ref. Reference Nathan202). These processes, together with lipid oxidation, are proatherogenic, particularly in injured arteries in hypertension. Ang II has significant pro-inflammatory actions in the vascular wall, inducing the production of ROS, cytokines and adhesion molecules and activation of redox-sensitive inflammatory genes (Ref. Reference Suematsu203).
Ang II modulates expression of pro-inflammatory molecules in the vessel wall, including vascular cell adhesion molecule 1, intercellular adhesion molecule (ICAM) and E-selectin, through redox-dependent pathways (Refs Reference Pueyo204, Reference Kranzhöfer205). In VSMCs, Ang II stimulates VCAM-1 production, chemokine (C–C motif) ligand 2 (CCL2/MCP-1) and interleukin 6 (IL-6) (Ref. Reference Phillips and Kagiyama206), which stimulates recruitment of mononuclear leukocytes into the vessel media. Many of these factors are increased in plasma from hypertensive patients, and it has been suggested that elevated circulating levels of cytokines and chemokines might reflect vascular inflammation and target organ damage in hypertensive patients (Refs Reference Hlubocka207, Reference Malmqvist208).
In addition to VSMCs, circulating and tissue leukocytes, lymphocytes and macrophages produce cytokines, chemokines and other pro-inflammatory mediators in response to Ang II. The adaptive immune response has an important role in this. Various T-cell subsets, including T helper 1 (Th1) cells (which produce interferon γ), Th2 cells (which produce IL-4), Th17 (which produce IL-17) and T suppressor cells including regulatory T cells (Treg) (which express the transcription factor FOXP3), have crucial roles in the development of Ang-II-dependent hypertension and in the progression of vascular remodelling (Refs Reference Schiffrin209, Reference Guzik210, Reference Harrison211). Central and pressor effects of Ang II are critical for activation of T cells and development of vascular inflammation (Ref. Reference Harrison211). Th17 cells seem to be particularly important in Ang-II-induced hypertension (Refs Reference Marvar212, Reference Madhur213, Reference Hoch214, Reference Vinh215).
The role of AT2R in the process of vascular inflammation is unclear. C-reactive protein, which is an inflammatory marker and independent predictor of incident hypertension, causes a sustained increase in blood pressure in mice and is associated with reduced vascular AT2R expression (Ref. Reference Vongpatanasin216). Genetic polymorphism A1675G in the gene encoding AT2R reduces cardiovascular risk and the severity of atherosclerosis by modifying inflammation, especially in hypertensive males (Ref. Reference Tousoulis217). In isolated human monocytes, AT2R stimulation enhanced prostaglandin-E2-mediated increase in MMP1, which has been associated with atherosclerotic plaque instability (Refs Reference Kim, Zhou and Wahl218, Reference Versari219).
Ang II, vascular ageing and hypertension
The age-associated changes in blood vessels that occur in healthy individuals include increased arterial wall thickness, reduced compliance, increased stiffness and decreased lumen diameter, typical features of the hypertensive vascular phenotype (Refs Reference de Chaves and Narayanaswami220, Reference Scalera221). These structural changes are associated with impaired endothelial function, caused by decreased production of vasodilators, such as NO and prostacyclins, and increased bioavailability of ROS (Ref. Reference Scalera221). Consistent with a dysfunctional endothelium is increased vasoconstriction and decreased fibrinolysis. Arterial ageing is a predominant risk factor for the onset of cardiovascular disease, such as hypertension, and is associated with activation of the RAS, increased vascular stiffness, intima–media thickening, calcification and a pro-inflammatory phenotype (Refs Reference Scalera221, Reference Wang, Monticone and Lakatta222). Moreover, vascular repair systems become progressively impaired with ageing. These features are characteristic of the vascular phenotype in hypertension, and in fact hypertension is considered to be a critically important factor in accelerated ageing of the vasculature (Ref. Reference Min223). In hypertension, this process assumes a more rapid course, eventually resulting in premature cardiovascular disease, including stroke, myocardial infarction and peripheral artery disease as well as vascular dementia. Cellular and molecular mechanisms underlying age-associated changes of the vascular system are unclear, but cardiovascular cells, including stem and progenitor cells, undergo senescence. Senescent cells enter irreversible growth arrest, exhibit a flattened and enlarged phenotype, and express genes, such as negative cell cycle regulators p53 (TP53) and p16 (CDKN2A), that differ from those normally expressed (Refs Reference Camici224, Reference Kortlever225). Factors implicated in cellular senescence include decreased telomerase activity and telomere shortening, DNA damage and genomic instability. These processes are modulated by Ang II and are highly redox sensitive. Hence increased oxidative stress, in part due to increased RAS activation, is an important promoter of cellular senescence. In fact the free-radical theory of ageing was proposed over 50 years ago, when Harman postulated that ROS induce macromolecular oxidative modifications and oxidative damage (Ref. Reference Harman226). There are now extensive data indicating that ROS bioavailabilty and RAS activation are increased in ageing as well as in hypertension (Refs Reference Nilsson, Lurbe and Laurent227, Reference Andreassi228, Reference Min229). In support of this, aged mice treated with inhibitors of the RAS, including losartan (AT1R blocker) and epleronone (aldosterone receptor blocker), showed reduced cardiovascular fibrosis and arrhythmias compared with untreated counterparts; also, mice deficient in the Agtr1a gene encoding AT1AR showed a markedly prolonged lifespan, possibly due to an increased number of mitochondria and upregulation of the prosurvival gene sirtuin 3 (Sirt3) (Refs Reference Stein230, Reference Benigni231, Reference Cassis232, Reference Kim233). ATRAP may have a role in regulating AT1R-mediated vascular ageing. ATRAP, which normally reduces AT1R signalling with enhancement of receptor internalisation, attenuated AT1R-mediated vascular senescence through inactivation of the calcineurin–NFAT (nuclear factor of activated T cells) pathway (Ref. Reference Min61). Aldosterone, through mineralocorticoid receptors, has also been implicated in vascular ageing, in part through activation of ERK1/2 (Ref. Reference Krug234).
Ang II and vascular calcification
Vascular calcification is associated with mineralisation of the internal elastic lamina and elastic fibres within the media, resulting in stiffened vessels and increased pulse pressure. Arterial calcification is not uncommon in ageing, chronic kidney disease, diabetes, atherosclerosis and hypertension and is related to cardiovascular morbidity and mortality (Refs Reference Huybers and Bindels235, Reference Ishiyama236, Reference Rosito237). Vascular calcification is a tightly regulated process similar to bone formation (Ref. Reference O'Neill238). Factors that trigger and promote calcification include abnormalities in mineral metabolism, particularly hyperphosphataemia and hypercalcaemia (Refs Reference Freedman239, Reference Li and Giachelli240), that promote VSMC differentiation to an osteoblastic phenotype (Refs Reference Freedman239, Reference Li and Giachelli240). This is driven by upregulation of transcription factors such as CBFA1 (RUNX2) and MSX2, and bone morphogenetic protein 2 (BMP2), which are critically involved in normal bone development and control the expression of osteogenic proteins, including osteocalcin, osteonectin, alkaline phosphatase, collagen I and bone sialoprotein (Refs Reference Freedman239, Reference Li and Giachelli240, Reference Alam241). Another mechanism contributing to vascular mineralisation is loss of calcification inhibitors, such as fetuin-A, matrix Gla protein, pyrophosphate and osteopontin (Refs Reference Alam241, Reference Reynolds242). Molecular processes underlying this remain to be fully defined but vasoactive agents may be important modulators of vascular calcification. ET-1 and urotensin II promote arterial calcification, whereas adrenomedullin and C-type natriuretic peptide inhibit calcification (Refs Reference Aikawa243, Reference Li, Yang and Giachelli244). Recent evidence suggests that the RAS may also be important. Ang II influences calcification by redox-sensitive pathways that stimulate expression of BMP2 and CBFA1 (Refs Reference Armstrong245, Reference Tokumoto246), and through modulation of Ca2+ and Mg2+ transport through cation channels, such as TRPM7 (Refs Reference He247, Reference Montezano248). AT1R blockade can inhibit arterial calcification by disrupting vascular osteogenesis, suggesting that patients with vascular calcification may benefit therapeutically from ARBs.
Clinical implications
The RAS has a major role in the pathophysiology of clinical hypertension, diabetes, chronic kidney disease and heart failure. As such, pharmacological agents that block key components of the RAS, such as ACE inhibitors, Ang II receptor blockers and direct renin inhibitors, have gained wide clinical use for these indications. Despite the enormous advances in these therapies, there is still morbidity and mortality of patients treated with RAS inhibitors. The reasons for this are complex and relate, in part, to the fact that there is still a paucity of information on the molecular mechanisms whereby Ang II mediates effects in physiological and pathological conditions in humans. The present review highlights some new trends in Ang II biology that provide insights at the clinical level. In particular, emerging evidence indicates that Ang II may have a pathophysiological role in processes not previously considered to be Ang II dependent, such as immune activation, arterial calcification and vascular ageing. These conditions are becoming major clinical problems as the population ages, and if there is indeed an Ang-II-sensitive component, then inhibition of the RAS might have important clinical potential beyond the pathologies for which ACE inhibitors and ARBs are classically used. In addition, identification of new components of the RAS, such as the (pro)renin receptor, Ang-II-derived peptides [e.g. Ang-(1-7)] and AT2R, provides possibilities for the development of new therapeutic strategies, some of which are already in clinical use such as direct renin inhibitors, aldosterone synthase inhibitors and AT2R agonists. Understanding the basic science of the RAS and how Ang II signals through its specific receptors, particularly in humans rather than in experimental models, should facilitate the discovery of new therapeutic targets to better manage patients with cardiovascular and kidney disease.
Expert opinion
Structural alterations and endothelial dysfunction of resistance arteries are hallmarks of hypertension. Initial factors contributing to this process involve increased transmural pressure, changes in blood flow and endothelial dysfunction. The subsequent alterations of VSMC growth, migration, differentiation, calcification, production of extracellular matrix proteins and inflammation, stimulated in large part by activation of the RAS, are then responsible for the resulting vascular remodelling. Of the numerous factors influencing remodelling in hypertension, Ang II is especially important. Over the recent past, our views of Ang II have changed; rather than being thought of as a simple vasoconstrictor, it is now seen as a complex growth factor that mediates effects through diverse signalling pathways involving PLC/PKC/Ca2+, PLA2, PLD, MAPKs, tyrosine kinases, proto-oncogene expression, RhoA/Rhokinase, inflammation and cell cycle modulation. It has also become clear that the RAS is not a simple endocrine organisation but rather a complex organisation involving a rich tissue RAS, multiple Ang-II-generating enzymes, many biologically active Ang-II-derived peptides and numerous receptors that are regulated by GIPs. Through increased generation of ROS and activation of redox-sensitive transcription factors, Ang II promotes expression of cell adhesion molecules and induces synthesis of pro-inflammatory mediators and growth factors. Some of these events may involve factors derived from microparticles and EPCs. These molecular and cellular processes facilitate increased vascular permeability, leukocyte recruitment, calcification and vascular fibrosis, leading to vascular injury, structural remodelling and premature ageing – the vascular phenotype in hypertension. Targeting some of these molecular events with novel therapeutic strategies that would regress or prevent arterial remodelling and ageing may provide important vascular protection in hypertension and other forms of cardiovascular and age-related diseases.
Acknowledgements and funding
Studies performed by R.M.T. were supported by grants 57786 and 44018 from the Canadian Institutes of Health Research (CIHR). R.M.T. is supported through a Canada Research Chair/Canadian Foundation for Innovation award. A.C.M. is supported through a fellowship from the CIHR. D.B. is supported through a KRESCENT (Kidney Research Scientist Core Education and National Training Program) fellowship from the Kidney Foundation of Canada. The authors thank the peer reviewers for their constructive comments.




