Introduction
Intrauterine growth restriction (IUGR) remains a leading cause of perinatal mortality and neonatal morbidity worldwide. Survivors of IUGR are at increased risk of cardiovascular and metabolic disease in later life.Reference Barker 1 In particular, a mismatch between the prenatal and postnatal environments predisposes towards the development of an adverse metabolic profile.Reference Hanson and Gluckman 2 Individuals born small who demonstrate ‘catch-up’ growth are at highest risk of adult-onset diseases (reviewed by OngReference Ong 3 ), whereas the absence of catch-up growth appears to be protective.Reference Hofman, Cutfield and Robinson 4 It appears that early-life nutrition can influence gene expression, which can alter baseline metabolic function and thus determine vulnerability to later life exposures. Although the mechanisms by which the early-life environment mediates these gene expression changes is not completely understood, it is believed to be due at least in part to epigenetic modifications that alter gene expression in the absence of any change in the genomic DNA sequence.
The most widely studied epigenetic mechanism to date is DNA methylation, which occurs at specific palindromic sequences where a cytosine (C) residue is directly followed by a guanine (G), termed CpG dinucleotides. CpG dinucleotides occur throughout the genome but predominate in short stretches of G:C-rich and CpG-dense regions termed CpG islands, which are often (but not always) found in or near the promoter regions of genes. The C residues within CpG dinucleotides exhibit variable degrees of methylation that differ from cell to cell and tissue to tissue. The presence of a methyl group inhibits the attachment of transcription factors.Reference Kass, Pruss and Wolffe 5 Accordingly, hypermethylation (particularly of the promoter region of a gene) is often associated with reduced gene expression or gene silencing, whereas relative hypomethylation is associated with enhanced gene expression. By contrast, intragenic DNA methylation may be associated with active transcription. Genome-wide DNA methylation studies comparing monozygotic with dizygotic human twin pregnancies have highlighted the important contribution that the intrauterine environment makes to the neonatal epigenome, even amongst genetically identical individuals.Reference Gordon, Joo and Powell 6 Various rat models of IUGR are characterized by hypermethylation of genes including insulin-like growth factor (IGF)1 and the glucocorticoid receptor (NRC31), and hypomethylation of the insulin receptor (INSR) in the offspring between 3 weeks and 18 months of life.Reference Fu, Yu, Callaway, Lane and McKnight 7 – Reference Zeng, Gu, Liu and Huang 10
The overnourished adolescent sheep model is characterized by placental and fetal growth restriction and premature delivery relative to the normally developing pregnancies of adolescents receiving a control dietary intake.Reference Wallace, Luther and Milne 11 The prenatal insult leads to a 42% reduction in uteroplacental blood flow,Reference Wallace, Milne, Matsuzaki and Aitken 12 which limits nutrient supply to the fetus, resulting in hypoglycaemia and hypoinsulinaemia by late pregnancy.Reference Wallace, Milne, Aitken and Hay 13 , Reference Wallace, Regnault, Limesand, Hay and Anthony 14 Historically, the degree of uteroplacental compromise is such that ∼50% of overnourished pregnancies result in significant IUGR (defined as a birth weight >2 s.d. below the mean birth weight of normally grown control fetuses), whereas the remaining 50% exhibit relatively ‘normal’ birth weight, despite the fact that they receive the same maternal nutritional manipulation and are similarly born, on average, 2–3 days preterm.Reference Wallace, Aitken, Milne and Hay 15
Relative to the ‘normal’ birth weight lambs of overnourished adolescent dams, IUGR lambs demonstrate increased fractional growth rate (FGR) to weaning for anthropometric parameters including weight, height and abdominal girth, although absolute catch-up growth is not seen.Reference Wallace, Milne, Aitken and Adam 16 Consequently, IUGR lambs still demonstrate lower carcass weight at weaning. Nevertheless, evidence of an adverse metabolic profile is evident from 7 weeks of age, when fasting glucose is increased and insulin secretion in response to glucose challenge is decreased.Reference Wallace, Milne, Aitken and Adam 16
We hypothesized that the placentally mediated fetal growth constraint that results in reduced birth weight and the aforementioned altered postnatal phenotype is, at least in part, epigenetically mediated. Therefore, the aim of the present study was to examine putative epigenetic changes underlying the observed differences in fractional growth between IUGR and normal birth weight lambs during early postnatal life. Given that nutrition per se may influence epigenetic status,Reference Begum, Stevens and Smith 17 , Reference Lan, Cretney and Kropp 18 we chose to compare IUGR v. normal birth weight offspring of exclusively overnourished adolescent ewes, as opposed to normal birth weight lambs of control-fed adolescent ewes, as this presents a unique opportunity to examine the effects of prenatal growth status independently of maternal nutrition during pregnancy and lactation and variations in gestation length. We chose a panel of candidate genes with potential relevance to postnatal growth and metabolism and for which ovine genomic sequences were already published. We hypothesized that IGF1 methylation, in particular, would be affected by prenatal growth restriction as differences in circulating IGF1 levels are present in the offspring of overnourished relative to control-intake adolescent ewes in fetal (0.9 gestation)Reference Wallace, Bourke and Aitken 19 and neonatal life: 13.8 v. 10.2 ng/ml, respectively, P<0.05, n=14 each (Wallace JM, unpublished data).
Materials and methods
Experimental animals
Animal procedures were approved by the UK Home Office under the Animals (Scientific Procedures) Act 1986 and by local ethics committee review. Ewes were housed in individual pens under natural lighting conditions at the Rowett Institute of Nutrition and Health (57°N, 2°W). Embryos were harvested on day 4 post-oestrus from 12 superovulated donor ewes (Scottish Blackface×Border Leicester, ∼2.5 years old) that had been inseminated by a single sire (Dorset Horn) on day 0. Embryos were transferred in singleton into the uteri of 65 oestrus-synchronized adolescent recipient ewes (Dorset Horn×Mule, ∼8.5 months old) to establish exclusively singleton pregnancies of precisely known gestational age and maximum genetic homogeneity, as previously described.Reference Wallace, Da Silva, Aitken and Cruickshank 20 Pregnancy rate was determined by transabdominal ultrasound at 45 days gestation, at which stage viable pregnancies were confirmed in 44 ewes (68%). Three ewes went on to miscarry before 90 days’ gestation (term=145 days). The remaining 41 pregnancies continued uninterrupted and were allowed to result in spontaneous vaginal delivery (see below).
Nutritional management
Following an initial 3-day re-alimentation period after embryo transfer, steadily increasing amounts of a complete diet were offered gradually over the next 2 weeks until the level of daily food refusal was ∼15% of the total offered (equivalent to ad libitum intakes). The level of food offered was reviewed three times per week and individually adjusted on the basis of daily food refusal rates throughout pregnancy. The complete diet provided 12 MJ of metabolizable energy and 140 g of crude protein/kg and was offered in two equal rations at 8 am and 4 pm (see Wallace et al.Reference Wallace, Milne, Redmer and Aitken 21 for details of diet composition and analyses). Following parturition, all ewes continued to be fed to appetite to maximize milk yield. Lambs had access to their mothers’ feed throughout the 11-week lactation and males remained gonad intact.
Parturition management
From 135 days gestation (the earliest point commensurate with live birth in the overnourished adolescent paradigm), ewes were supervised 24 h a day and allowed to deliver spontaneously. A standardized proactive regime of neonatal care applied to all lambs (including resuscitation at birth and prophylactic antibiotic coverage) was used in view of the otherwise potentially very high rates of neonatal mortality in IUGR lambs (up to 62%) owing to prematurity and impaired passive immunity and/or low nutrient intake secondary to inadequate colostrum supply.Reference Wallace, Aitken and Cheyne 22 Lambs were dried and weighed at the time of birth. After delivery, 10 IU intravenous oxytocin (Intervet UK Ltd) was administered to ewes in order to induce milk let down. The udder was stripped by hand and the total volume of colostrum was determined before being fed back to the lamb by bottle or feeding tube. Lambs require at least 50 ml colostrum/kg birth weight to acquire sufficient antibody protection.Reference Wallace, Milne and Aitken 23 If maternal colostrum yield fell below this minimum requirement then the difference was provided to the lamb in the form of pooled donor colostrum that had been collected and frozen previously. Lambs were weighed at regular intervals during the neonatal period to determine whether any further supplementary feeds were required to ensure lamb survival.
Blood sampling and analysis
Venous blood was sampled at the time of birth and on the day of necropsy (see below) and immediately centrifuged at 2000 g for 20 min at 4°C. IGF1 and insulin levels were quantified in duplicate by radioimmunoassay, as previously described.Reference Wallace, Milne, Aitken and Adam 16
Necropsy and tissue sampling
Three lambs (two IUGR and one normal) died or were euthanized for welfare reasons during the neonatal period, which left 38 surviving lambs available for further study. Lambs were weighed at 5-day intervals until weaning at 11 weeks of age in order to determine absolute and FGRs. This time point was chosen, as the adverse postnatal phenotype that is characteristic of this IUGR model is established by this stage, including altered growth trajectory, body conformation and glucose metabolism.Reference Wallace, Milne, Aitken and Adam 16 Overall, FGR (%/day) was calculated by expressing the live weight gain between birth and necropsy at 77.5±0.4 days gestation as a proportion of lamb birth weight and dividing by the time interval between birth and necropsy. Thereafter, all lambs were humanely killed by intravenous injection of pentobarbital sodium (20 ml) and underwent complete postmortem examination. All major internal organs were examined macroscopically and weighed. Samples of hepatic tissue from the same position and same lobe in each animal were snap frozen in isopentane chilled with liquid nitrogen and stored at −80°C, pending DNA and RNA extraction.
DNA extraction and pyrosequencing
From each lamb, 25 mg of hepatic tissue was lysed and homogenized, and DNA was extracted using the DNEasy Blood & Tissue Kit (Qiagen, Uppsala, Sweden). The DNA concentration of each sample was determined using a ND-1000 spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA) and samples were run on an Agilent 2100 Bioanalyser to exclude RNA contamination. In order to validate the pyrosequencing reactions, a fully methylated control was prepared by treating ovine genomic DNA with CpG methylase, M.SssI (EC 2.1.1.37; Zymo Research Corporation, Irvine, CA, USA) and a fully unmethylated control was prepared using a modified genome-wide amplification approach with the GenomiPhi™ DNA Amplification Kit (GE Healthcare, Little Chalfont, Bucks, UK) and DNA polymerase derived from the bacteriophage φ29, as described.Reference Umetani, de Maat, Mori, Takeuchi and Hoon 24 From each test and control sample, 400 ng DNA was modified overnight with sodium bisulphite (which converts unmethylated C bases to uracil whilst leaving unmethylated ones intact) using the EZ DNA Methylation-Gold™ Kit (Zymo Research Corporation). All samples were bisulphite-converted in a single batch in order to avoid conversion bias.
Genomic DNA sequences for ovine genes of the somatotrophic axis were identified via the NCBI Nucleotide database. Principal genes associated with growth were searched for amongst all published genomic DNA sequences for Ovis aries (28,512 at time of writing). If an ovine sequence was available for a particular gene of interest, this was analysed using Methyl Primer Express® v1.0 (Applied Biosystems, Foster City, CA, USA) to determine the presence or absence of one or more C:G islands according to the following criteria: length >200 bp; GC content >55%; observed to expected C:G ratio >0.65. A total of 24 CpG islands were identified in the following 10 genes: insulin (INS), IGF1 and 2, H19, growth hormone (GH), INSR, insulin-like growth factor 1 receptor (IGF1R), insulin-like growth factor 2 receptor (IGF2R), growth hormone receptor (GHR) and glucocorticoid receptor (NRC31), detailed in Table 1. H19 was included because, although it does not itself code for a protein, DNA methylation of this gene is inversely correlated with IGF2 expression.Reference Gao, Suppola and Liu 25 Sequences were subsequently modified to account for the widespread degradation of C to T (outwith CpG dinucleotides) that occurs during bisulphite conversion and highlight CpGs as sites of interrogation. Primers for polymerase chain reaction (PCR) and pyrosequencing were designed specifically on the bisulphite-converted sequences using PyroMark Assay Design software v2.0.1.15 (Qiagen). Care was taken to avoid the inclusion of CpG dinucleotides within the PCR primer sequences in order to prevent amplification bias, while optimizing the number of CpGs within the sequencing primer to maximize the number of sites at which DNA methylation could be quantified (see supplementary data file). Forward/reverse primers need to be 9–40 bases in length (optimum 20), have a melting temperature of 58–60°C and a GC content of 30–60%, avoid runs of identical nucleotides (<3 sequential Gs) and have no >2 Gs and/or Cs within the final five nucleotides at the 3′ end. Sequencing primers needed to have a melting temperature of 68–70°C, avoid any Gs at the 5′ end and be located as close as possible to the other two primers without overlapping. A total of 14 assays were designed covering a total of 57 CpG sites across the 10 genes of interest. Subsequently, PCR reactions were set up using biotinylated primers (Table 2) and the AmpliTaq Gold® DNA Polymerase Low DNA Kit (Applied Biosystems) and run on a PTC-225 Peltier Thermal Cycler (MJ Research, Watertown, MA, USA). After verifying that an amplicon of the expected length had been generated using gel electrophoresis, PCR products were sequenced in a PSQ 96MA pyrosequencer using Pyromark Gold Q96 Reagents (Qiagen) and custom-designed sequencing primers (Table 3). All 38 samples were processed on a single plate for each gene of interest. At each of the 57 individual CpG sites examined, percentage DNA methylation was calculated as the ratio of C to T, reflecting the proportion of methylated v. unmethylated DNA in the original sample.
Table 1 Details of 24 CpG islands identified within 10 somatotrophic axis genes

C, cytosine; G, guanine; INS, insulin; IGF2, insulin-like growth factor 2; IGF1, insulin-like growth factor 1; GH, growth hormone; GHR, growth hormone receptor; INSR, insulin receptor; IGF1R, insulin-like growth factor 1 receptor; IGF2R, insulin-like growth factor 2 receptor; DMR, differentially methylated region.
a No accession number available for INSR – sequence taken from McGrattan et al.Reference McGrattan, Wylie and Bjourson 70
Table 2 PCR primers for 14 assays interrogating CpG sites in 10 genes of interest

PCR, polymerase chain reaction; INS, insulin; IGF2, insulin-like growth factor 2; IGF1, insulin-like growth factor 1; GH, growth hormone; IGF1R, insulin-like growth factor 1 receptor; IGF2R, insulin-like growth factor 2 receptor; GHR, growth hormone receptor; INSR, insulin receptor.
Table 3 Sequencing primers for 14 assays interrogating CpG sites in 10 genes of interest

PCR, polymerase chain reaction; INS, insulin; IGF2, insulin-like growth factor 2; IGF1, insulin-like growth factor 1; GH, growth hormone; IGF1R, insulin-like growth factor 1 receptor; IGF2R, insulin-like growth factor 2 receptor; GHR, growth hormone receptor; INSR, insulin receptor.
RNA extraction and real-time reverse transcription PCR (RT-PCR)
Messenger RNA (mRNA) levels for four of the above 10 genes (IGF1, IGF2, IGF1R and IGF2R) were determined by quantitative RT-PCR. For each lamb, ∼25 mg of hepatic tissue was lysed and homogenized and RNA was extracted using the RNeasy Mini Kit (D-40724; Qiagen), quantified and quality checked via capillary electrophoresis using the Agilent 2100 Bioanalyser. Next, ∼30 ng total RNA from each sample was reverse transcribed in triplicate to complementary DNA using TaqMan reverse transcription reagents and MultiScribe reverse transcriptase (Applied Biosystems, Warrington, Cheshire, UK). Ovine-specific probes and primer sets were designed using Primer Express® and are detailed in Table 4. Polymerization and amplification reactions for each RT were performed in duplicate using a 7500 Fast Real-Time PCR system (Applied Biosystems) at 60°C for 40 cycles. Samples were randomized to ensure that each prenatal growth category and both genders were equally represented in each of four 96-well PCR plates. In addition, a quality control sample generated from a RNA pool was run on each plate and used to calculate the inter- and intra-assay coefficient of variation (cov) for each gene of interest. Intra-plate cov varied from 3.4 to 5.9% for individual genes (overall mean±s.e.m.=4.3±0.57%), whereas inter-plate cov varied from 2.03 to 11.9% (overall mean±s.e.m.=6.2±2.08%). The individual sample mRNA expression for each gene of interest was expressed relative to the sample’s own internal 18S RNA content using 18S PDAR kit reagents (Applied Biosystems).
Table 4 RT-PCR primers and probes for four somatotrophic axis genes of interest

RT-PCR, reverse transcription polymerase chain reaction; IGF1, insulin-like growth factor 1; IGF2, insulin-like growth factor 2; IGF1R, insulin-like growth factor 1 receptor; IGF2R, insulin-like growth factor 2 receptor.
a Nucleotide sequences for ovine-specific genes were obtained from the National Center for Biotechnology Information (NCBI) database.
Data analysis
There was a continuous distribution of birth weights and lambs were classified as IUGR based on a 2 s.d. cut-off below the mean birth weight of normally grown lambs born to control-fed adolescent ewes in earlier studies using this model.Reference Wallace, Aitken, Milne and Hay 15 , Reference Wallace, Aitken and Cheyne 22 Accordingly, the threshold for IUGR was <4000 g and the remaining lambs were classified as non-IUGR. In view of anticipated gender differences, data are additionally presented by sex. Although we had no direct control over the gender of the embryos transferred, the eventual male to female ratio amongst the lambs was well balanced and supported this approach. After confirming normality and equality of variance using Q-Q plots and Levene’s test, respectively, the four groups (normal male, normal female, IUGR male and IUGR female) were compared using general linear model (GLM) in order to assess the effects of IUGR status, gender and their potential interactions. In circumstances where GLM indicated strong effects of either prenatal growth status or gender, data were additionally combined, irrespective of gender (i.e. all IUGR v. all normal birth weight lambs) and IUGR status (all males v. all females) and compared using Student’s t-test. Percentage DNA methylation was compared between groups at each individual CpG site and as an average for each individual assay and gene of interest. Correlations were assessed using Pearson’s product moment test. All data are presented as mean±s.e.m., unless otherwise stated. Formal statistical significance was considered to have been reached where P<0.05.
Results
Anthropometric and biochemical parameters at birth and necropsy
Table 5 details anthropometric and biochemical parameters at the time of birth and at necropsy at ∼77 days of age, presented by gender and dichotomous IUGR status. There were no significant differences in the requirements for or indeed the duration of supplementary feeding between lambs classified as IUGR or ‘normal’ birth weight (P>0.05, data not shown). IUGR lambs were, on average, 32% lighter at birth than their normally grown counterparts (3.26±0.14 v. 4.80±0.12 kg, P<0.001, respectively, for males and females combined). The average weight of the entire cohort was 4.11 kg. In spite of increased FGR relative to normal lambs (11.0±0.38 v. 8.5±0.22%/day, P<0.001), absolute catch-up growth was not observed within the first 11 weeks of life and prenatally growth-restricted lambs remained ∼13% lighter at the time of necropsy (live weight: 31.1±1.02 v. 35.8±0.77 kg, P<0.001). Although there were no significant gender differences in birth weight, by 11 weeks of age males were significantly heavier than females (35.7±0.98 v. 31.5±0.81 kg, P=0.002 for IUGR and normal birth weight combined). The reduced liver weight attributable to both IUGR status and female gender was largely proportionate and was no longer significantly different from normal birth weight and/or male lambs when expressed in relative (i.e. empty body weight specific) terms (P>0.05). At birth, plasma concentrations of both IGF1 and insulin were reduced (P<0.02) in IUGR lambs (in keeping with reduced fetal nutrient supply) and were independent of gender. However, at necropsy the influence of IUGR status on IGF1 levels was reversed, with modestly but significantly higher plasma concentrations in the prenatally growth-restricted lambs (P<0.05). Furthermore, irrespective of growth category, male lambs had markedly higher circulating IGF1 levels than female lambs (597±30 v. 380±18 ng/ml, P<0.001 from GLM and by t-test). Peripheral insulin concentrations at necropsy were also higher in male v. female lambs (P<0.05) and were independent of prenatal growth status. Moderate variability was noted in plasma insulin levels, which likely reflects the fact that these were not fasted samples, given that lambs were kept together with their mothers until point of necropsy to prevent separation anxiety.
Table 5 Selected anthropometric and biochemical parameters at the time of birth and necropsy at 77.5±0.5 days postnatal age

IUGR, intrauterine growth restriction; FGR, fractional growth rate; IGF1, insulin-like growth factor 1.
Significant P values (<0.05) are highlighted in bold.
Hepatic DNA methylation
Table 6 shows the mean percentage methylation for each of the 14 assays investigating a total of 10 different genes of interest in relation to postnatal growth and metabolism, whereas Table 7 details the methylation status at each individual CpG site. There were no statistically significant differences between IUGR and normal birth weight lambs for any of the genes examined, although IGF2R methylation tended to be greater in IUGR v. normal birth weight lambs (54.3±0.47 v. 53.2±0.41%, P=0.087), independent of gender. In contrast, independent of IUGR status, IGF1 exons 2 and 3 methylation was increased in female v. male lambs (75.5±0.14 v. 74.6±0.26%, P=0.01; and 90.1±0.19 v. 89.3±0.29%, P<0.05, respectively, from GLM and by t-test) and consequently overall IGF1 gene methylation was significantly greater in females (86.2±0.14 v. 85.3±0.22%, P=0.008), independent of IUGR status. Further, overall hepatic IGF1 methylation was negatively correlated with IGF1 plasma levels at necropsy (r=−0.455, n=38, P=0.005), irrespective of gender.
Table 6 Mean DNA methylation of 10 somatotrophic axis genes (presented by exon where applicable) in hepatic tissue at 77.5±0.5 days postnatal age

IUGR, intrauterine growth restriction; INS, insulin; IGF1, insulin-like growth factor 1; IGF2, insulin-like growth factor 2; GH, growth hormone; INSR, insulin receptor; IGF1R, insulin-like growth factor 1 receptor; IGF2R, insulin-like growth factor 2 receptor; GHR, growth hormone receptor.
Significant P values (<0.05) are highlighted in bold.
Table 7 DNA methylation of 50 individual CpG sites across 10 somatotrophic axis genes in hepatic tissue at 77.5±0.5 days postnatal age

IUGR, intrauterine growth restriction; INS, insulin; IGF1, insulin-like growth factor 1; IGF2, insulin-like growth factor 2; GH, growth hormone; INSR, insulin receptor; IGF1R, insulin-like growth factor 1 receptor; IGF2R, insulin-like growth factor 2 receptor; GHR, growth hormone receptor.
NRC31 methylation is not shown by individual CpG site as minimal detectable methylation at several loci prevented meaningful comparisons between groups.
Significant P values (<0.05) are highlighted in bold.
Hepatic mRNA expression
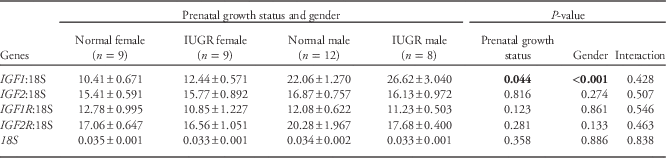
Table 8 shows the relative mRNA expression of IGF1, IGF2, IGF1R and IGF2R in hepatic tissues obtained at necropsy. In keeping with the plasma IGF1 results detailed above, IGF1 mRNA expression was marginally greater in IUGR relative to normal birth weight lambs (P<0.05), and was markedly higher in males v. females (23.9±1.48 v. 11.4±0.49, P<0.001 from GLM and by t-test), independent of prenatal growth status. Irrespective of gender or growth status, mRNA expression of IGF1 was positively correlated with plasma IGF1 levels at necropsy (r=0.884, n=38, P<0.001) and negatively correlated with overall hepatic IGF1 methylation (r=−0.507, n=38, P=0.002). There was no impact of prenatal growth status or gender on hepatic IGF2, IGF1R and IGF2R mRNA abundance.
Table 8 mRNA expression of the insulin-like growth factors (IGF) and their receptors relative to 18S in hepatic tissue at 77.5±0.5 days postnatal age
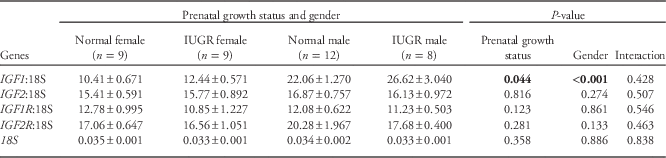
mRNA, messenger RNA; IUGR, intrauterine growth restriction; IGF1R, insulin-like growth factor 1 receptor; IGF2R, insulin-like growth factor 2 receptor.
Significant P values (<0.05) are highlighted in bold.
Associations between molecular parameters and phenotype
Figure 1 summarizes the gender differences seen in three different aspects of IGF1 gene function, namely protein levels (a), mRNA expression (b) and DNA methylation (c). In addition to the strong relationships between these three parameters in the expected and biologically meaningful direction, IGF1 methylation was negatively correlated with carcass weight (r=−0.460, n=38, P=0.005) and plasma insulin levels (r=−0.580, n=38, P<0.001) at necropsy. By contrast, H19 methylation was positively correlated with carcass weight at necropsy (r=0.494, n=38, P=0.002). These relationships were independent of lamb gender. In addition, IGFR1 and GH methylation were inversely correlated with lamb birth weight (r=−0.524, n=38, P=0.018) and plasma IGF1 levels at necropsy (r=−0.535, n=38, P=0015), respectively, in male lambs only, whereas IGF2 methylation was positively correlated with plasma insulin levels at necropsy (r=0.519, n=38, P=0.027) in female lambs only, irrespective of IUGR status.

Fig. 1 Plasma insulin-like growth factor 1 (IGF1) levels (a) plus hepatic mRNA expression (b) and DNA methylation of the IGF1 gene presented by gender at 77.5±0.5 days postnatal age in 38 lambs born to overnoursished adolescent ewes and characterized as normal birth weight (n=21, open bars) or IUGR (n=17, closed bars).
Discussion
Effects of IUGR
In the present study, IUGR lambs were noted to have significantly higher IGF1 mRNA expression and higher circulating IGF1 protein levels relative to normal birth weight lambs. This represented a switch from the pattern observed at the time of birth, when IGF1 concentrations were lower in IUGR lambs, most likely reflecting reduced fetal nutrient supply. This reversal of the IGF1 differential by 11 weeks postnatal life and markedly altered absolute and fractional growth velocity during the early neonatal period are in keeping with the phenomenon of neonatal catch-up growth, which occurs in the event of a mismatch between the pre- and postnatal environmentsReference Gluckman and Hanson 26 and is often attributed to putative epigenetic changes. However, in the present study, despite a major differential in birth weight and markedly altered rates of growth, prenatal growth restriction had no significant impact on the methylation status of 10 genes variously involved in somatic growth and metabolism. A statistical trend towards higher IGF2R methylation was noted in IUGR lambs (P=0.087) relative to normally grown lambs, which could well represent a chance observation, especially as it was not accompanied by any measurable change in IGF2R mRNA expression. Alternatively, however, it may reflect an antecedent effect of IUGR during intrauterine life. Unlike IGF1R, which mediates the mitogenic effects of IGF1 and IGF2, IGF2R is a clearance receptor that antagonizes IGF2 action, and disruption of the IGF2R gene results in increased serum and tissue levels of IGF2 and fetal overgrowth.Reference Ludwig, Eggenschwiler and Fisher 27 Relative hypermethylation of the IGF2R gene at the particular site examined, which is an imprinted differentially methylated region, would hypothetically be associated with reduced gene expression, which would serve to maximize IGF2 availability. The fact that IGF2 is predominant during fetal but not neonatal life might potentially explain the failure to demonstrate differences in IGF2R expression at 11 weeks of age, at which time persistent differences in DNA methylation may simply represent stigmata of earlier events in the life course. IGF2R hypermethylation has been reported previously in children referred to geneticists for short stature following IUGR.Reference Turner, Mackay and Callaway 28
It was interesting to observe striking yet consistent differences in the overall degree of methylation between genes (e.g. ∼90% for INS v. <2% for GHR) and within genes (e.g. ∼4% at a single locus in IGF1 exon 2 compared with >85% for all others tested). The GHR had the lowest overall methylation, yet clearly maintained the potential to become heavily methylated, as a 94% methylation status was achieved in the control sample treated with M.SssI. The region examined was just proximal to exon 1B, which is expressed in multiple tissues and contains a putative promoter.Reference Adams 29 In addition, NRC31 was noted to have no measurable methylation at many loci, and minimal degrees of methylation were detectable. The degree of NRC31 methylation observed herein is consistent with that reported in the hypothalamus of prenatally undernourished sheep, which ranges from 0.3 to 0.6% during fetal lifeReference Begum, Stevens and Smith 17 to ∼1% at 5 years of age.Reference Begum, Davies and Stevens 30
In general, the apparent lack of a measurable epigenetic effect of IUGR following overnourishment of the pregnant adolescent ewe suggests that no permanent alterations in somatotrophic gene function are associated with this prenatal insult, which is reassuring. The lack of apparent epigenetic changes may relate to the timing of the insult in our model, given that the onset of IUGR is relatively late, being confined to the final third of gestation.Reference Carr, Aitken, Milne, David and Wallace 31 Although the impact of IUGR is arguably greatest in late gestation, when energy demand is maximal, there is accumulating evidence that the epigenome is more susceptible at an earlier window of development. Initial studies on individuals prenatally exposed to the Dutch famine of 1944–1945 reported reduced methylation of IGF2 Reference Heijmans, Tobi and Stein 32 and increased methylation of interleukin 10 (IL10), ATP-binding cassette subfamily A member 1 (ABCA1), maternally expressed 3 (non-protein coding) (MEG3), leptin (LEP) and GNAS antisense RNA (GNASAS)Reference Tobi, Lumey and Talens 33 only when the exposure occurred around the time of conception and not during late gestation. The same group subsequently tested for differences at the same loci in a separate cohort of adults born preterm after IUGR and found no differences in DNA methylation of IGF2, GNASAS, INSIGF and LEP relative to appropriate birth weight controls.Reference Tobi, Heijmans and Kremer 34 The latter results suggest that not all long-term morbidity of IUGR resulting from programming in utero is mediated by changes in DNA methylation and may involve other epigenetic mechanisms such as histone modificationsReference Munshi, Shafi, Aliya and Jyothy 35 or microRNAs (miRNAs).Reference Chuang and Jones 36 For example, IUGR induced by bilateral uterine artery ligation in the rat has been demonstrated to modify the histone code of the IGF1, PPAR-γ and NRC31 genes,Reference Fu, Yu, Callaway, Lane and McKnight 7 , Reference Ke, Schober and McKnight 37 , Reference Joss-Moore, Wang and Baack 38 and levels of miR132.Reference Joss-Moore, Wang and Ogata 39 Alternatively, non-epigenetic insults on tissue structure and function may be responsible for long-term adverse effects on health. For example, IUGR in the rat is characterized by decreased pancreatic β-cell mass and islet vascularity leading to impaired insulin secretion,Reference Gatford and Simmons 40 whereas in the human low birth weight is associated with reduced nephron numbersReference Ritz, Amann, Koleganova and Benz 41 and exaggerated sympathetic nerve activity during adulthood,Reference Boguszewski, Johannsson, Fortes and Sverrisdottir 42 both of which are implicated in the prenatal programming of hypertension. Given that there was a measurable impact of IUGR on IGF1 mRNA expression and circulating IGF1 in the present study, both of which were increased relative to normal birth weight lambs, it remains possible that alternative epigenetic or indeed non-epigenetic mechanisms may be responsible.
Effects of gender
In the present study, irrespective of prenatal growth status, male lambs had significantly higher IGF1 mRNA expression and higher circulating IGF1 protein levels compared with female lambs. No gender differences were present at the time of birth, suggesting that this sexual dimorphism in IGF1 emerges during the neonatal period, reflected by serial IGF1 samples over the first 11 weeks of life.Reference Wallace, Milne, Aitken and Adam 16 Hepatic IGF1 mRNA expression and IGF1 protein levels were each inversely correlated with hepatic IGF1 DNA methylation, which was significantly greater in female v. male lambs at 11 weeks postnatal age. Relative IGF1 hypomethylation in males is consistent with increased transcription and hence greater IGF1 protein levels. However, absolute differences between male and female lambs were ultimately very small at <1%, which raises questions about their biological significance. Notably, however, these gender differences were highly statistically significant, were present at five separate CpG sites and occurred in the expected direction of effect relative to the clear differences in protein and mRNA expression. The chances that differences in three separate aspects of IGF1 gene function were detectable by chance are infinitely small; however, it is accepted that DNA methylation changes alone are unlikely to explain the total variation in IGF1 mRNA and protein expression observed in this cohort of lambs. Notably, the small differential between groups (<1%) is similar to previous reports of DNA methylation in humans across a wide range of genes, including male v. femaleReference Hall, Volkov and Dayeh 43 and type 2 diabetic v. non-diabetic comparisons.Reference Gu, Gu, Hilding, Ostenson and Brismar 44 These studies have also revealed similar variance with respect to DNA methylation (<1%) among human subjects, despite the arguably much greater degree of genetic heterogeneity when compared with our highly controlled sheep model. Furthermore, in the aforementioned studies of hypothalamic NRC31 DNA methylation, a small decrease of only 0.6–0.3% was associated with a five-fold increase in mRNA expression.Reference Begum, Stevens and Smith 17 , Reference Begum, Davies and Stevens 30
We found that the differences in IGF1 methylation were confined to exons 2 and 3. The lack of similar changes in exon 4 may reflect the fact that its mRNA is not expressed at all in sheep.Reference Dickson, Saunders and Gilmour 45 In support of this assumption, methylation proximal to exon 4 was greater than at any region studied, suggesting minimal gene expression or gene silencing. Interestingly, the putative promoter site for IGF1 is located just proximal to exon 1 but is reportedly not GC rich;Reference Ohlsen, Dean and Wong 46 therefore, it is unsurprising that no conventional CpG island was found at this location in the published ovine sequence. Given that the aims of the present study were to examine DNA methylation in CpG islands, we did not measure methylation of this putative promoter, which is a potential limitation of our approach. Moreover, as the regions of IGF1 investigated were all intragenic, it is possible that differential methylation here may have effects that are different to promoter methylation.Reference Maunakea, Chepelev, Cui and Zhao 47 To our knowledge, gender differences in IGF1 methylation have not previously been reported in any species, although other genes have been shown to be differentially methylated in males v. females. For example, the aforementioned studies on survivors of the Dutch Hunger Winter found that effects on INS, IGF and LEP were restricted to men and that changes in GNASAS were more pronounced in women. Moreover, IGFR2 methylation in normal (unexposed) controls was 2.6% higher in men v. women in adult life.Reference Tobi, Lumey and Talens 33 IGFR1 methylation was higher in male db/db mice (homozygous for a point mutation in the leptin receptor) compared with female db/db mice and both male and female controls.Reference Nikoshkov, Sunkari and Savu 48 In general, human epigenetic studies have shown that CpG sites generally show greater methylation in males, with the exception of imprinted genes, in which DNA methylation appears to be more equal between sexes.Reference El-Maarri, Becker and Junen 49 Consequently, the relative hypomethylation of IGF1 seen here in males is novel but certainly in keeping with recognized gender differences in circulating IGF1 in the young lamb.Reference Wallace, Milne, Aitken and Adam 16 Studies in other species have similarly demonstrated elevated IGF1 levels in males v. females at various stages of the life course. For example, peripheral IGF1 concentrations are higher in male mice during early puberty,Reference Callewaert, Sinnesael, Gielen, Boonen and Vanderschueren 50 whereas in the rat no measurable differences occur until well into adult life, at around 12 weeks of age.Reference Fukuda, Usuki and Mukai 51 In humans, IGF1 levels tend to be higher in boys than girls between 6 and 18 years of age;Reference Xu, Gu and Pan 52 however, no significant differences in IGF1 are apparent in early adulthoodReference Gatford, Heinemann and Thompson 53 or old age,Reference Taekema, Ling and Blauw 54 despite persistent gender differences in body composition and glucose metabolism in both humans and our sheep model of IUGR.Reference Wallace, Milne, Aitken and Adam 16 , Reference Wallace, Aitken and Cheyne 22 , Reference Wallace, Milne, Adam and Aitken 55 In females, there were clear associations between lower circulating IGF1 levels, body weight and radial/ulnar growth rates, and higher adiposity, leptin levels and thoracic growth rates; however, to what extent this sexual dimorphism is governed by differences in DNA methylation remains unclear. As discussed above in relation to IUGR status, it remains possible that alternative epigenetic mechanisms are responsible for the observed differences at the mRNA/protein level. For example, gender differences have been reported in histone acetylationReference Tsai, Grant and Rissman 56 and miRNA regulation,Reference Sharma and Eghbali 57 some of which may be mediated by differences in sex steroids. Although circulating levels of oestrogen and testosterone are likely to have been low at the point of necropsy several weeks before puberty, these steroid hormones could nevertheless have impacted pituitary GH secretion resulting in altered hepatic IGF1 action. Clinical studies have demonstrated associations between hypogonadism and low GH and precocious puberty and high GH,Reference Meinhardt and Ho 58 and exposure to sex steroids during neonatal life can impact the GH secretory pattern later in life by modulating the number of GH releasing hormone neurones in the hypothalamus.Reference Chowen, Frago and Argente 59 In addition, exposure to xenoestrogens during neonatal life alters GH-dependent liver proteinsReference Ramirez, Bourguignon and Bonaventura 60 and oestrogen replacement has been associated with reduced circulating IGF1 levels in adult women.Reference Ho, Gibney, Johannsson and Wolthers 61 Accordingly, a direct or indirect effect of reproductive steroids on the IGF1 system cannot be ruled out and may interact with or indeed eclipse the relatively small epigenetic changes observed herein.
Strengths and weaknesses
Bisulphite sequencing methods provide very high-resolution assessment of methylation at specific loci, but are limited by the fact that only a small number can be examined in any one reaction. Furthermore, the specific sites are limited to those around which functional primer sets can be designed, which are largely dependent upon the neighbouring gene sequences. Finding primers that are specific enough and do not form secondary (e.g. hairpin or dimer) structures when working with bisulphite-converted DNA is a major challenge, as the standard four-base genetic code is massively simplified to one comprising just three bases. In addition, the highly stringent primer specifications (detailed in the Materials and methods section) greatly limited the number of viable sets for each (relatively short) gene sequence, greatly restricting the sites within the genomic DNA that could be examined. Clearly, it remains possible that changes outwith the 57 CpG sites studied could have been missed. Array-based technologies have been developed for the human and mouse that can simultaneously examine up to 450,000 CpG sites in a single assay; however, no such commercial kits are currently available for sheep. More recently, the role of 5-hydroxymethylcytosine (5hmC), which is a marker of so-called ‘active demethylation’ in which methylated C is oxidized by ten-eleven translocation enzymes, has been attracting increasing attention, especially in the field of cancer epigenetics.Reference Wang, Tang, Lai and Zhang 62 Unfortunately, the methodology used herein does not currently distinguish between 5-methylcytosine and 5hmC; therefore, we are unable to comment on any potential influence of demethylation, and any future studies should take this into account. It is also a potential limitation that the methodology used cannot recognize differences in methylation between the maternal and paternal alleles, called imprinting, which is known to influence a number of different genes including H19. This may arguably be less important here, given the use of a single sire, which completely controls for paternal genetics.
A further limitation is that, although accepted criteria were used to identify CpG islands within the available ovine gene sequences, the biological significance of differential DNA methylation at these specific sites has largely not been assessed. Most of the ovine genes investigated herein were originally sequenced with the aim of examining their exonic arrangement rather than focussing on the 5′ untranslated regions (proximal introns) where the majority of CpG islands are known to lie.Reference Kass, Pruss and Wolffe 5 As the DNA methylation assays developed herein are novel, direct comparisons with the limited number of previous sheep studies are not possible. Sinclair et al.Reference Sinclair, Allegrucci and Singh 63 reported alterations in DNA methylation in sheep fetuses following periconceptual manipulation of maternal dietary vitamin B and methionine content at 4% of 1400 loci in a gender-specific manner using restriction landmark genome scanning; however, the identity of these loci was not explicitly stated. Wang et al.Reference Wang, Zhang and McMillen 64 examined methylation of IGF2/H19 and IGF2R in the late fetal and early postnatal heart using combined bisulphite restriction analysis, but found no significant changes secondary to ovine IUGR induced by carunclectomy. Begum et al.Reference Begum, Stevens and Smith 17 and Lan et al.Reference Lan, Cretney and Kropp 18 recently demonstrated altered methylation status of NRC31 and the proopiomelanocortin gene, and IGF2R and H19, respectively, in a variety of ovine fetal tissues following maternal dietary manipulations (underfeeding and variable energy source, respectively), but did not report any associated impact on fetal weight or mRNA expression of the same panel of genes. Direct comparisons with other ovine studies may also be limited by the fact that the present study used donor superovulation, which is known to influence DNA methylation of imprinted genes including H19 Reference Market-Velker, Zhang, Magri, Bonvissuto and Mann 65 and IGF2.Reference Fortier, McGraw and Lopes 66 As all animals herein underwent the same assisted reproductive techniques and oocytes from 9 of 11 donors ultimately produced both IUGR and normal birth weight lambs with a balanced ratio of males to females, it is unlikely that this limits comparisons between groups. The use of a single sire also limited any variation in paternal genetics.
It should also be noted that we only examined DNA methylation in a single tissue and at a single time point. Liver was chosen for this study as most published work on the epigenetic impact of IUGR has been carried out using this tissue type, and because the liver is an important metabolic organ that plays a central role in the regulation of postnatal growth. However, ultimately epigenetic changes observed in one tissue cannot necessarily be extrapolated to others.Reference Herzog, Galvez and Roks 67 For example, adipose tissue would be a good candidate for further investigation into the influence of IUGR and gender on early-life metabolism. As we only examined animals at weaning, it is unknown if sexual dimorphism in IGF1 methylation is present at the time of birth, or indeed whether it persists into adult life. Hence, it remains unclear whether differential IGF1 methylation is programmed in utero or represents dissimilarities that emerge alongside other sex differences such as fasting metabolite levels and relative adiposity during postnatal growth and development. In support of this concept, there is evidence that epigenetic changes can occur and can be prevented during postnatal life. For example, amelioration of catch-up growth by dietary manipulation in growth-restricted rat pups appears to prevent changes in IGF1 methylation that otherwise emerge in the first few weeks,Reference Tosh, Fu and Callaway 68 and is associated with an improved metabolic profile.Reference Lim, Armitage, Stefanidis, Oldfield and Black 69 Consequently, it is possible that the changes seen herein simply represent gender differences in ontogeny of the somatotrophic axis, rather than a ‘fetal programming’ effect per se.
Conclusion
In summary, IUGR induced by overnourishment of pregnant adolescent ewes did not significantly impact DNA methylation of key growth axis genes, but gender differences in live weight and body conformation in early postnatal life were associated with sexual dimorphism in hepatic DNA methylation and mRNA expression of the IGF1 gene and plasma IGF1 concentrations. Future work should focus on more targeted methylation analysis around putative promoter and regulatory regions of IGF1 and evaluate for possible changes in 5hmC.
Acknowledgements
The authors would like to thank Chaz Mein and Eva Wozniak at the Genome Centre, Queen Mary University of London, who were integral to the development of the DNA methylation assays and processing of samples.
Financial Support
This work was supported by Wellbeing of Women (D.J.C., grant number RTF318) and the Scottish Government’s Rural and Environment Science and Analytical Services Division including the Strategic Partnership for Animal Science Excellence (J.M.W., R.P.A., J.S.M. and C.L.A.).
Conflicts of Interest
None.
Ethical Standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guides on the care and use of laboratory animals (sheep) and has been approved by the institutional committee (Rowett Institute local ethics review committee).
Supplementary Material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/S2040174415001415