Introduction
The ‘dual’ symbiotic nature of lichens was first proposed in the late 19th century by Simon Schwendener (Reference Schwendener1868). However, the validity of his hypothesis was contested, often fervently, for almost another hundred years by those who saw lichens as whole organisms rather than a symbiosis between fungi and ‘algae’ (Chlorophyta or Cyanobacteria) (Honegger Reference Honegger2000). The concept of lichens as a mycobiont/photobiont symbiosis is now universally accepted (see Nash Reference Nash2008); however, debate still continues as to the true nature of this relationship, including whether it represents a controlled parasitism or mutualism. A bacterial contribution to lichen nutrition (e.g., via N2-fixation) has also been considered for quite some time (Henckel & Yuzhakova Reference Henckel and Yuzhakova1936), and contemporary culture-independent studies are increasing our understanding of diverse populations of bacteria associated with lichens and their potential functional roles within the symbiosis (Cardinale et al. Reference Cardinale, Vieira de Castro, Müller, Berg and Grube2008; Grube et al. Reference Grube, Cardinale, Vieira de Castro, Müller and Berg2009; Hodkinson & Lutzoni Reference Hodkinson and Lutzoni2009; Bates et al. Reference Bates, Cropsey, Caporaso, Knight and Fierer2011; Hodkinson et al. in press; reviewed in Grube & Berg Reference Grube and Berg2009).
In addition to algal and lichenized fungal symbionts, numerous other eukaryotic organisms are known to associate with lichens, which they use for food or shelter (Gerson Reference Gerson1973). Some moth species, for example, preferentially lay their eggs under the protective covering of lichens (Thomson 1958), and oribatid mites are known to burrow into lichens on which they feed (Grandjean Reference Grandjean1950). Members of diverse invertebrate groups including arthropods, rotifers, nematodes, and tardigrades have all been isolated from lichen thalli where they presumably inhabit internal areas (Stubbs Reference Stubbs1989; Bartels & Nelson Reference Bartels and Nelson2007). Lichens also host numerous fungal species, in addition to the mycobiont, such as lichenicolous (lichen-associated) fungi (Lawrey & Diederich Reference Lawrey and Diederich2003; Lawrey et al. Reference Lawrey, Binder, Diederich, Molina, Sikaroodi and Ertz2007) and endolichenic fungi (which grow within the interior of lichens) (Girlanda et al. Reference Girlanda, Isocrono, Bianco and Luppi-Mosca1997; Suryanarayanan et al. Reference Suryanarayanan, Thirunavukkarasu, Hariharan and Balaji2005; U'Ren et al. Reference U'Ren, Lutzoni, Miądlikowska and Arnold2010). Interestingly, recent phylogenetic studies suggest lichen thalli themselves may be ‘cradles’ of fungal diversification in the Ascomycota (Arnold et al. Reference Arnold, Miądlikowska, Higgins, Sarvate, Gugger, Way, Hofstetter, Kauff and Lutzoni2009).
Despite a growing body of literature on organisms associated with lichens, we still have limited knowledge of the extent of eukaryotic diversity that may be associated with individual lichen thalli. Here we carried out a preliminary survey of eukaryotes associated with surface-sterilized foliose lichen thalli, using high-throughput pyrosequencing with eukaryotic-specific primers targeting the 18S rRNA gene. As an extensive diversity of bacteria has been shown to be associated with internal surfaces of lichens (Cardinale et al. Reference Cardinale, Vieira de Castro, Müller, Berg and Grube2008; Grube et al. Reference Grube, Cardinale, Vieira de Castro, Müller and Berg2009; Bates et al. Reference Bates, Cropsey, Caporaso, Knight and Fierer2011; B. P. Hodkinson et al. in press), we also compared our eukaryotic diversity results with previously published analyses of bacterial diversity recovered from the same specimens.
Materials and Methods
Sampling
To minimize the influence of eukaryotic organisms haphazardly found on rock surfaces in our survey, we selected thalli of three umbilicate (i.e., attached to the substratum only at a single, central point), foliose green algal lichen species. Rhizoplaca chrysoleuca, Umbilicaria americana, and Umbilicaria phaea, were collected at a site in northern Colorado (40·01°N, 105·47°W) from rock outcrops. Each thallus was removed from the rock substratum using a sterile knife and placed into individual sterile plastic collection bags. The specimens were then transported back to the laboratory on ice and processed immediately.
Surface sterilization and isolation of community DNA
Each lichen thallus was surface sterilized using the most thorough protocol outlined by Arnold et al. (Reference Arnold, Miądlikowska, Higgins, Sarvate, Gugger, Way, Hofstetter, Kauff and Lutzoni2009), to avoid amplification of DNA originating from eukaryotic organisms that may have only come into contact with external surfaces of our specimens. The sterilization procedure was as follows: samples were washed for ∼30 s in sterile ultrapure laboratory-grade (Milli-Q) water to remove debris from outer surfaces; they were then immersed and agitated separately in 96% ethanol for 10 s, followed by 0·5% NaOCl (bleach) for 2 min, and 70% ethanol for 4 min. A small piece of the lichen thallus with the approximate dimensions of 2 cm2 was removed from the sample immediately after surface sterilization for use in genomic DNA extraction. We used the commercially available PowerSoil DNA isolation kit (MoBio Laboratories, Carlsbad, CA) to extract genomic DNA from thalli after an initial treatment to enhance DNA yield. This treatment included separately grinding each sample with a sterile pestle and mortar under liquid N2, placing the macerated samples into individual 2-ml bead-beating tubes with kit buffer, and immersion in a 65°C water bath for 10 min. After heating, extractions proceeded according to the kit protocol.
PCR amplification of 18S rRNA genes and bar-coded pyrosequencing
Extracted genomic DNA representing communities of lichen-associated organisms were prepared for pyrosequencing following the protocol outlined by Fierer et al. (Reference Fierer, Hamady, Lauber and Knight2008), using only eukaryotic-specific primers. Briefly, the method includes targeted PCR amplification of a portion (up to ∼600 bp) of the 18S small subunit rRNA gene, triplicate PCR product pooling (per sample) to mitigate reaction-level PCR biases, and pyrosequencing using the eukaryotic-specific primer set F515 (5′-GTGCCAGCMGCCGCGGTAA-3′) and R1119 (5′-GGTGCCCTTCCGTCA-3′). We have demonstrated in silico that this primer set should amplify 18S rRNA genes from a broad range of eukaryotic groups with few biases (see Supplementary Fig. S1). The F515 primer included a Roche FLX+ pyrosequencing adapter (Roche Applied Science, Indianapolis, IN) and a 2-bp linker sequence (GT), and R1119 incorporated 12-bp bar-coded sequences (each unique to an individual specimen), an AG linker, and a FLX+ sequencing adapter (Roche). The PCR was carried out in 25 µl reaction mixtures, containing 1 µl (5 µM [each]) of forward and reverse primers, 10 µl of 5Prime Hot master mix (Eppendorf-5Prime, Gaithersburg, MD), and 12 µl MoBio PCR water. Each reaction mix received 1 µl of genomic community DNA as a template, and the following cycling parameters were used: 35 cycles (94°C for 45 s, 45°C for 30 sec, and 72°C for 1·5 min) were performed after an initial denaturation at 94°C for 3 min. Triplicate reaction mixtures per specimen were combined and purified using an UltraClean PCR cleanup kit (MoBio), followed by quantification using PicoGreen dsDNA (Invitrogen, Carlsbad, CA). The bar-coded PCR products from all samples were normalized in equimolar amounts in a pooled sample, and sent for sequencing at Roche Applied Science.
For pyrosequencing, 1 µl of PCR product was checked for quality with a Bioanalyzer 2100 DNA 1000 chip (Agilent Technologies, Santa Clara, CA), quantified using the Quant-iT PicoGreen assay (Invitrogen), and then diluted to 1 × 107 molecules per µl. The PCR products were clonally amplified using the GS FLX Titanium LV emPCR Kit (Lib-A) following a modified version of the manufacturer's instructions (emPCR Amplification Method Manual: Lib-L LV, GS FLX+ Series: XL+). Briefly, amplicons were immobilized onto DNA-capture beads, micro-reactors were formed of a single DNA-containing bead in an emulsion oil sphere, and emulsified samples were subjected to PCR amplification on 96-well plates. After amplification, emulsions were chemically broken and beads carrying the amplified DNA library were recovered and washed, and then DNA-positive beads were purified using a biotinylated primer/streptavidin-coated magnetic beads complex. Recovered library beads were melted from magnetic beads yielding a population of bead-bound single-stranded DNA templates. After sequencing primer annealing, beads were counted with a Mulitsizer 3 (Beckman Coulter, Brea, CA), two million enriched beads from each of two amplicon pools were loaded into one of two regions in a 70 × 75 mm PicoTiterPlate device, and then subjected to 400 cycles of modified pyrophosphate-based DNA sequencing on a GS FLX+ automated sequencer following the manufacturer's protocol (Sequencing Method Manual, GS FLX+ Series: XL+ Kit, Roche). In this study, amplicons were sequenced from a single direction; however, the ‘A’ Adaptor (GS FLX Titanium LV emPCR Kit: Lib-A) is commonly employed for bi-directional amplicon sequencing. Our primer selection combined with GS FLX+ technology provided read lengths (up to ∼600 bp) at a more than sufficient resolution for the accurate taxonomic classification of micro-organismal sequences (Liu et al. Reference Liu, Lozupone, Hamady, Bushman and Knight2007).
Sequence processing
The QIIME software pipeline (Caporaso et al. Reference Caporaso, Kuczynski, Stombaugh, Bittinger, Bushman, Costello, Fierer, Gonzalez Peña, Goodrich and Gordon2010) was used to process raw sequence data, perform quality control, sample grouping (via the unique 12 bp bar-codes), phylotype binning, and taxonomic assignment of sequence data. Taxonomy for all eukaryotic phylotypes (sequences sharing ≥ 97% similarity) recovered from our lichen specimens was assigned in QIIME using BLAST (Altschul et al. Reference Altschul, Madden, Schäffer, Zhang, Zhang, Miller and Lipman1997), based on sequences from the SILVA comprehensive ribosomal RNA database (http://www.arb-silva.de/). Sequences representing the phylotypes were further screened for chimeras, which were removed from the dataset, and phylotypes assigned as ‘environmental samples’ in the SILVA database were subjected to BLASTn (Altschul et al. Reference Altschul, Madden, Schäffer, Zhang, Zhang, Miller and Lipman1997) searches in GenBank (http://www.ncbi.nlm.nih.gov/genbank/) to determine their taxonomic affiliation. Subsequent diversity analyses were performed using R statistical software (http://www.r-project.org/).
Results and Discussion
Eukaryotic survey
On average, 1008 eukaryotic sequences (from 862, 1021, to 1141) were recovered from our lichen samples. In addition to phylotypes representing the photo- (all 99·5% matches with Trebouxia) and mycobionts (>99% matches with Rhizoplaca or Umbilicaria) of each lichen species, a total of 50 other distinct eukaryotic phylotypes were recovered from the 2904 high-quality sequences generated by pyrosequencing of 18S rRNA genes associated with our specimens. These phylotypes represented diverse eukaryotic taxa from nine phyla within four major clades of Eukarya: Alveolata (Ciliophora), Fungi (Ascomycota, Basidiomycota, Blastocladiomycota, Chytridiomycota), Metazoa (Rotifera, Tardigrada), Rhizaria (Cercozoa), and Viridiplantae (Chlorophyta).
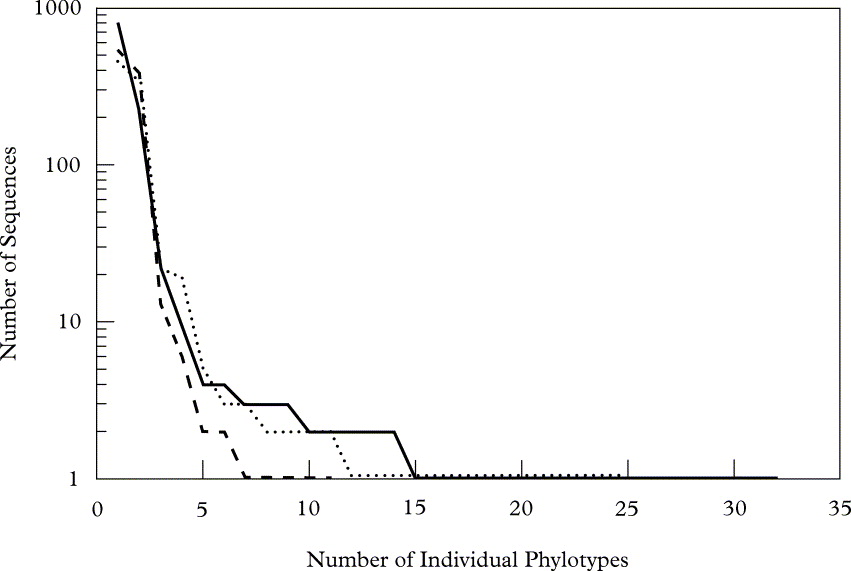
The relative abundance of each eukaryotic phylotype varied considerably within each specimen, with relative abundances characterized by an ‘L-shape’ curve (Fig. 1) that is typical for communities of micro-organisms (Fuhrman Reference Fuhrman2009) and indicates that the majority of phylotypes were relatively rare. These phylotypes essentially represented three levels of abundance: high (representing between 19% and 70% of the sequences recovered from each sample), medium (between 7% and 0·5%), and low (less than 0·5%). Invariably the high abundance group comprised two phylotypes representing the photo- and mycobionts. Three phylotypes were recovered in each lichen species that represented the mid-level group, and all but one (a 99·7% match to a taxon in Tardigrada) of these phylotypes corresponded to fungal taxa within the Ascomycota. The numerous low abundance phylotypes, many of these being ‘singletons’, represented only 1–4% of all sequences recovered from each lichen sample. Although many of these phylotypes matched taxa that are known to be associated with lichens (e.g., lichenicolous fungi, protozoa, rotifers and tardigrades), others represented taxa that are not (e.g., chytrids and hypogeous fungi).
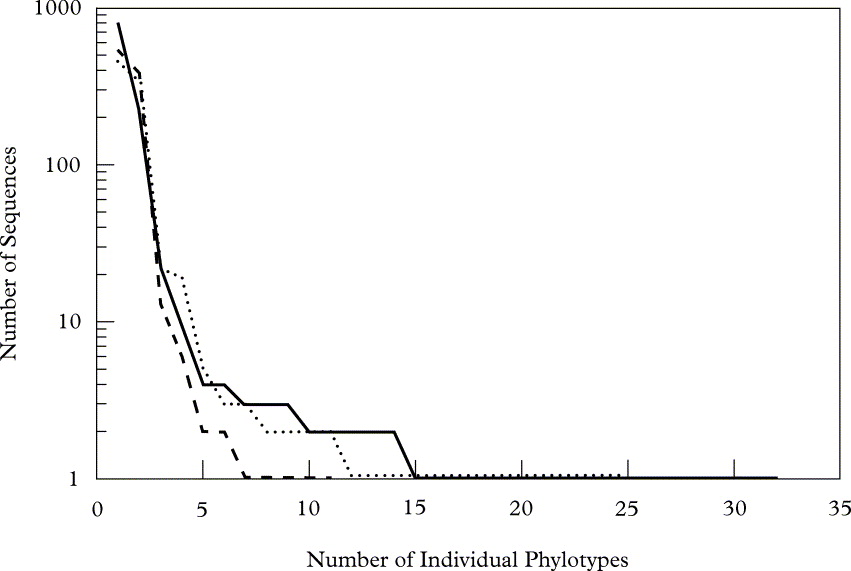
Fig. 1. Rank abundance graph of unique eukaryotic phylotypes recovered from each lichen species. The number of individual sequences of a given phylotype is shown on the y-axis (log scale) and the x-axis represents the ranked order of the phylotypes obtained from the lichens sampled (· · · · · R. chrysoleuca, —— U. Americana, - - - - - U. phaea).
Dominant phylotypes
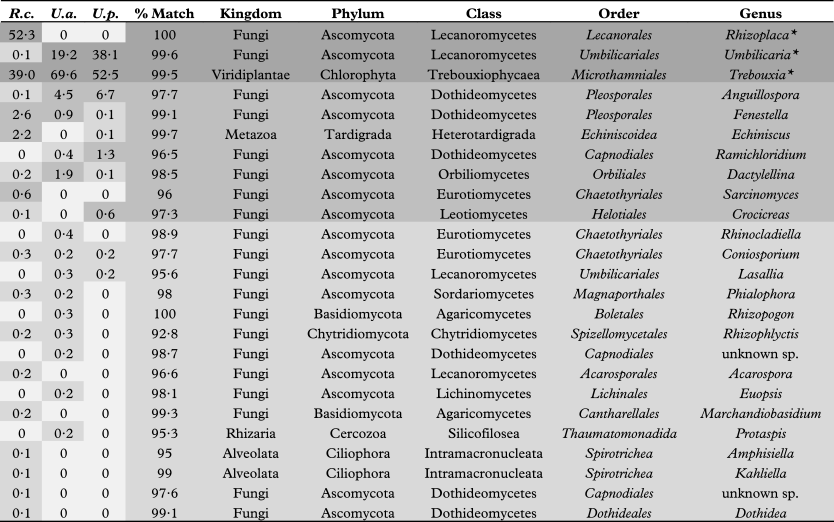
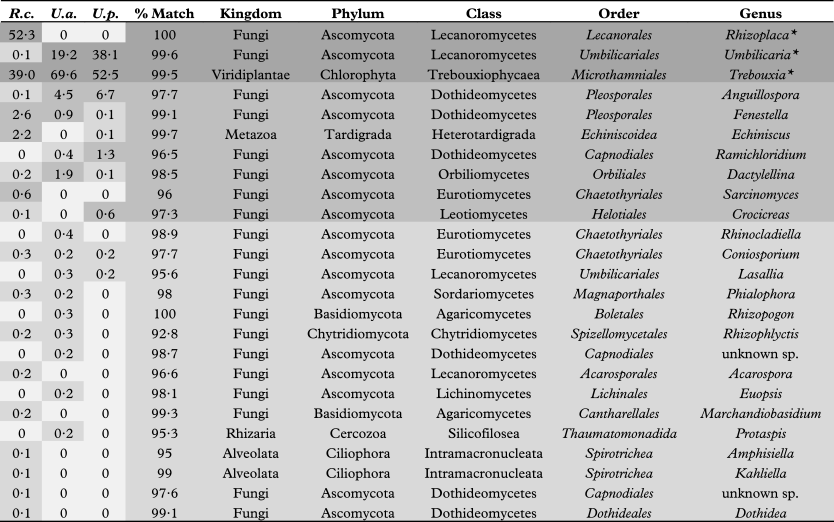
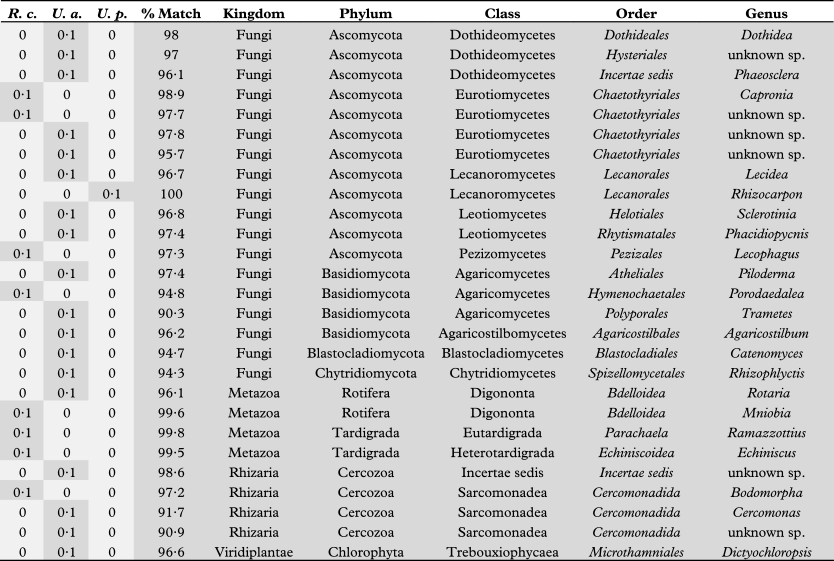
Table 1 shows the abundances for all phylotypes recovered in the survey indicating high, medium and low level abundance groups. Considering the large amount of photo- and mycobiont biomass in these samples, biont phylotype representation in the high abundance group is consistent with their corresponding 18S rRNA gene copies inherent in the sample DNA pool. Although the number of mid-level phylotype group sequences recovered was considerably lower than that of the high-level group (see Fig. 2), they do represent quantities of biomass that were significant enough to be detected over the biont 18S signature. Tardigrades of the order Echiniscoidea are known to inhabit lichens (Bartels & Nelson Reference Bartels and Nelson2007); therefore, the recovery of a phylotype corresponding to this taxon among our mid-level group is not surprising, as the organism or its eggs would constitute ample biomass to be detected in our relatively small (2 cm2) lichen sample. The other mid-level phylotypes represent several classes of ascomycetous fungi (Dothideomycetes, Eurotiomycetes, Leotiomycetes and Orbiliomycetes) that have been isolated from lichen thalli previously (Pfister & Liftik Reference Pfister and Liftik1995; Lawrey & Diederich Reference Lawrey and Diederich2003; Arnold et al. Reference Arnold, Miądlikowska, Higgins, Sarvate, Gugger, Way, Hofstetter, Kauff and Lutzoni2009), suggesting that this group corresponds to actual lichenicolous or endolichenic fungi inhabiting the thalli of our lichen samples. The low abundance group was the most diverse, representing all phyla as well as 82% of the unique phylotypes recovered in this study. Although some of these phylotypes may represent authentic lichen-associated organisms, others are probably only haphazardly present in our specimens. For example, some may be fungal remnants in the digestive tract of mycophagous invertebrates, such as tardigrades and bdelloid rotifers (Meininger & Spatt Reference Meininger and Spatt1988; Wilson & Sherman Reference Wilson and Sherman2010), which were also among the low abundance phylotypes recovered from our samples.
Table 1 Relative abundance (% of all 18S rRNA gene sequences in each sample) of phylotypes (* = bionts) recovered from the lichens species Rhizoplaca chrysoleuca (R.c.), Umbilicaria americana (U.a.), and Umbilicaria phaea, (U.p.), and their percent identity matches with eukaryotic taxa. High, medium, and low level abundance groups are indicated (darkest to lightest greys, respectively).
Comparison of eukaryotic and bacterial diversity
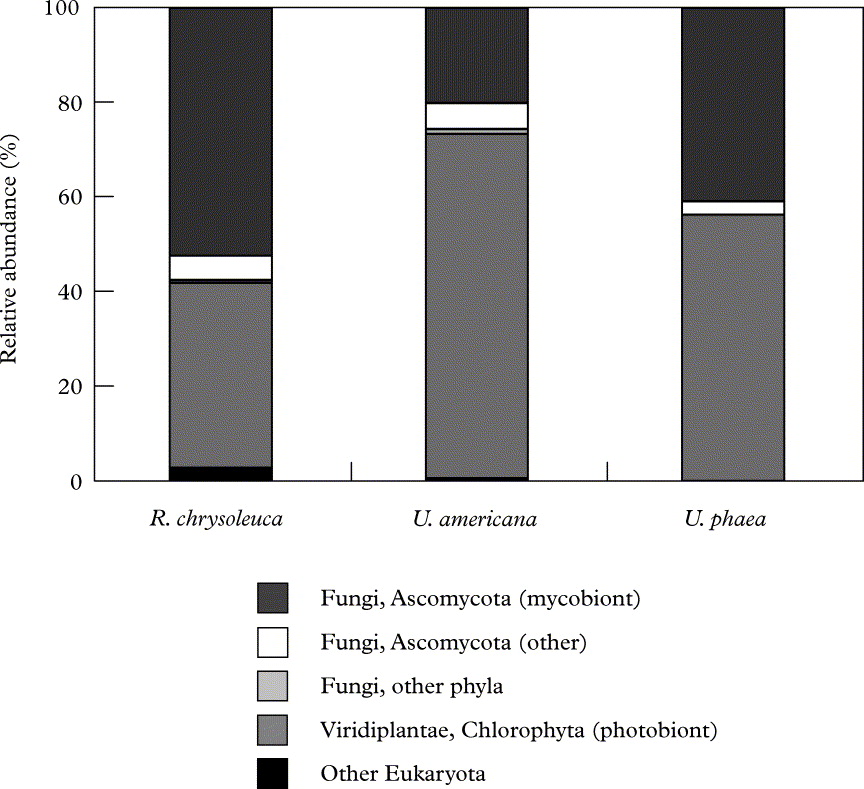
The high relative abundance of biont 18S copies in our lichen samples is also apparent in Figure 2, as is the fraction of non-biont ascomycetous taxa that probably represents many true lichen-inhabiting fungi. Overall, just two primarily biont-related phyla (Ascomycota and Chlorophyta) dominated our lichens, while the remaining phyla (Ciliophora, Basidiomycota, Blastocladiomycota, Chytridiomycota, Rotifera, Tardigrada and Cercozoa) had very low representation. In contrast, more higher order bacterial taxa, some with members probably having distinct functional roles in the lichen symbiosis (Grube & Berg Reference Grube and Berg2009; Hodkinson & Lutzoni Reference Hodkinson and Lutzoni2009; Bates et al. Reference Bates, Cropsey, Caporaso, Knight and Fierer2011; B. P. Hodkinson et al. in press), were noticeably present in these same lichen samples (Bates et al. Reference Bates, Cropsey, Caporaso, Knight and Fierer2011; see fig. 2 in that publication).
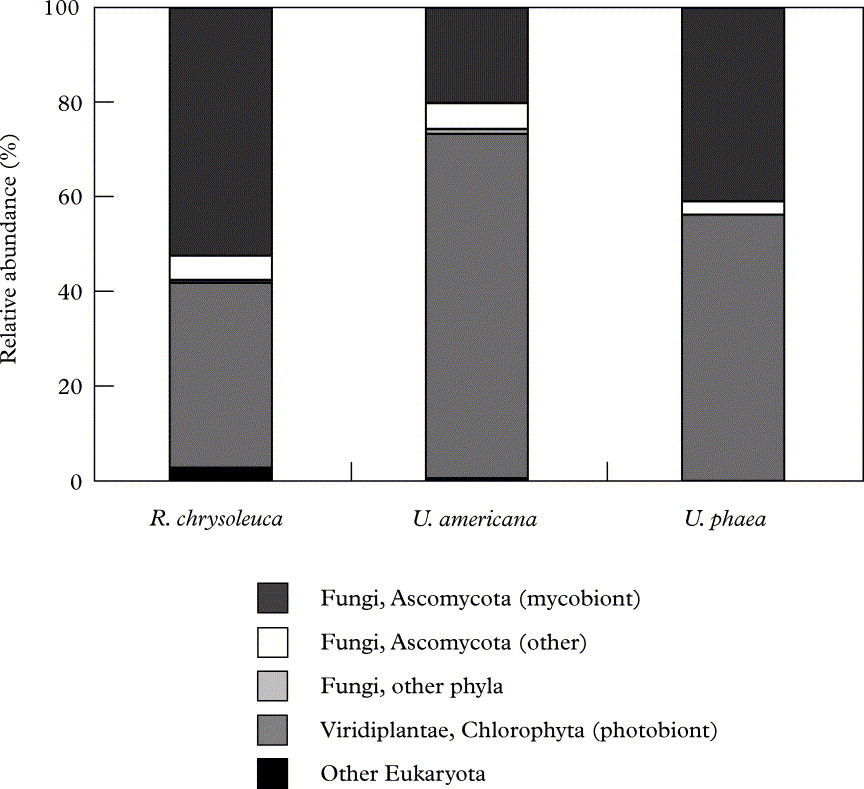
Fig. 2. Relative abundances of various major eukaryotic lineages recovered from each lichen species. Relative abundance was calculated as the percentage of sequences belonging to a particular lineage of all 18S rRNA gene sequences recovered from the lichens sampled.
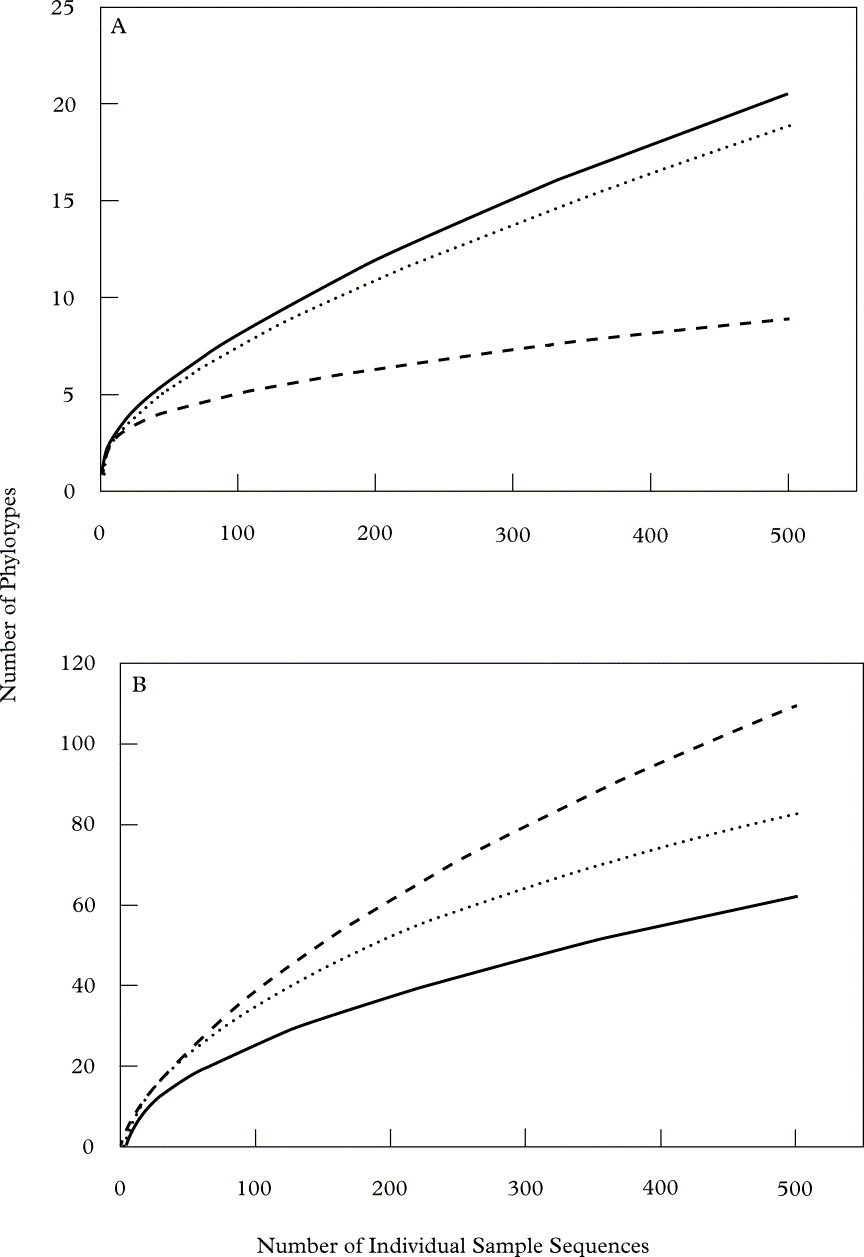
Rarefactionscurves (Fig. 3) highlight the considerable amount of micro-organismal diversity associated with even small fragments of lichen thalli, as plots for both eukaryotes and bacteria (see Bates et al. Reference Bates, Cropsey, Caporaso, Knight and Fierer2011) fail to asymptote. With the same sampling effort, however, eukaryotic diversity associated with lichens is more restricted than that of bacteria from the same lichen specimens. This may be attributed to the smaller size of bacteria compared to the eukaryotic organisms that our surveys suggest inhabit these samples, which may allow for more relative niche space for bacteria to occupy. Diversity values for our lichen samples were generally higher than those of previous DNA-based studies of endolichenic fungi (Arnold et al. Reference Arnold, Miądlikowska, Higgins, Sarvate, Gugger, Way, Hofstetter, Kauff and Lutzoni2009; U'Ren et al. Reference U'Ren, Lutzoni, Miądlikowska and Arnold2010), which may be attributable to overestimation by our pyrosequencing approach (Engelbrektson et al. Reference Engelbrektson, Kunin, Wrighton, Zvenigorodsky, Chen, Ochman and Hugenholtz2010). However, methodological differences make such comparisons difficult, and we were conservative in our method of phylotype binning in order to minimize inflation of diversity estimates. It is also interesting to note that the specimen with the highest level of eukaryotic diversity (Umbilicaria americana; Fig. 3A) also held the lowest level of bacterial diversity (Fig. 3B) and vice versa for U. phaea. This observation raises the question of possible competitive interactions between the eukaryotic (principally fungi) and bacterial populations that inhabit lichens; however the small number of lichens sampled here does not allow us to test this hypothesis. Conversely, the higher levels of diversity for U. americana may simply be the result of physical features of this species, such as the dense covering rhizomorphs on the lower surface of the thallus, which may provide excellent habitat for lichen associated eukaryotes.
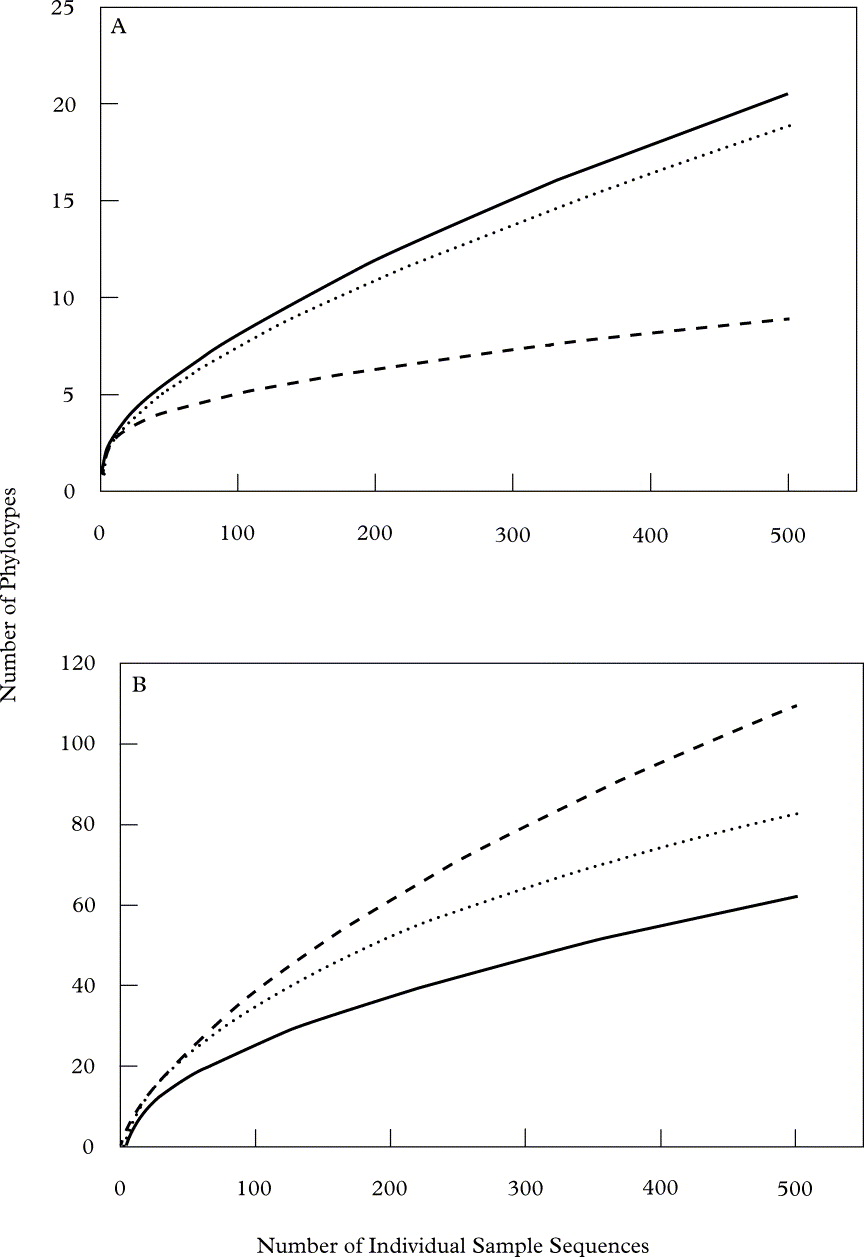
Fig. 3. Rarefaction curves depicting the richness (y-axis, as the number of unique phylotypes recovered) determined at an equal sampling effort (x-axis, the number of individual sequences recovered in each sample) of (A) eukaryotic and (B) bacterial (see Bates et al. Reference Bates, Cropsey, Caporaso, Knight and Fierer2011) phylotypes associated with each lichen species (· · · · · R. chrysoleuca, —— U. americana, - - - - - U. phaea).
Conclusions
Recent DNA-based studies continue to elucidate the extent of fungal (Lawrey et al. Reference Lawrey, Binder, Diederich, Molina, Sikaroodi and Ertz2007; Arnold et al. Reference Arnold, Miądlikowska, Higgins, Sarvate, Gugger, Way, Hofstetter, Kauff and Lutzoni2009; U'Ren et al. Reference U'Ren, Lutzoni, Miądlikowska and Arnold2010) and bacterial (Cardinale et al. Reference Cardinale, Vieira de Castro, Müller, Berg and Grube2008; Grube et al. Reference Grube, Cardinale, Vieira de Castro, Müller and Berg2009; Hodkinson & Lutzoni Reference Hodkinson and Lutzoni2009; Bates et al. Reference Bates, Cropsey, Caporaso, Knight and Fierer2011; B. P. Hodkinson et al. in press; reviewed in Grube & Berg Reference Grube and Berg2009) diversity that is present in lichen species. Although many invertebrate animals are known to associate with lichens (e.g., see Gerson Reference Gerson1973), this is the first culture-independent survey to document members of the Alveolata, Metazoa, and Rhizaria inhabiting internal surfaces of lichens. We have also contributed to the understanding of lichen-associated fungal diversity, recovering phylotypes corresponding to phyla (Blastocladiomycota and Chytridiomycota) of fungi not isolated previously from lichens. The presence of these fungal phylotypes, as well as those matching other fungal taxa that were relatively rare, serves as a caution for culture-dependent studies: lichen thalli may contain numerous potential fungal propagules that do not represent sizable biomass portions in lichens and are perhaps not true lichenicolous or endolichenic fungi.
Although the overwhelming abundance of biont related sequences in our survey may have limited our ability to resolve the full extent of eukaryotic diversity in our specimens, the sequences recovered in our pyrosequencing survey revealed diverse assemblages of organisms from several major eukaryotic clades which inhabit even small fragments of lichen thalli. This finding, along with the results of previous surveys of other lichen-associated micro-organisms, reinforces the concept that, in addition to being symbiotic systems where numerous symbiotic partners may interact, lichenscan also be considered minute ecosystems (Farrar Reference Farrar, Brown, Hawksworth and Bailey1976; Grube et al. Reference Grube, Cardinale, Vieira de Castro, Müller and Berg2009). Modern molecular tools are now allowing us to explore the intricacies of these systems in ways that were probably inconceivable to Schwendener and his contemporaries.
Members of the Fierer group helped in the laboratory and with the analyses. Garrett Cropsey assisted in sample collecting and processing. Laura Wegener Parfrey provided useful discussion and suggestions. We thank Mike Robeson and Bill Birky for assistance in locating eukaryotic SEM images, which were graciously provided by Byron Adams, Diego Fontaneto and Robert Roberson. Chinnappa Kodira of Roche Applied Science supervised the pyrosequencing. This research was funded by grants to RK and NF from the National Institutes of Health, the National Science Foundation, and the US Department of Agriculture.