


Introduction
Wild rice (Oryza rufipogon) is considered the ancestor of cultivated rice (Oryza sativa) and has been recognized as a precious germplasm resource for rice breeding (Sang and Ge, Reference Sang and Ge2013; Atwell et al., Reference Atwell, Wang and Scafaro2014). Dongxiang wild rice (DXWR), a strain of O. rufipogon found in Dongxiang County, China, has the most northerly habitat (28°14ʹN) among wild rice populations worldwide (Xie et al., Reference Xie, Agrama, Kong, Zhuang, Hu, Wan and Yan2010; Mao et al., Reference Mao, Yu, Chen, Li, Zhu, Xiao, Zhang and Chen2015). However, rapid urbanization around the habitat of DXWR has almost led to its extinction (Hu et al., Reference Hu, Wan, Li, Cai, Luo and Xie2012). Therefore, it should be effectively protected and exploited.
The discovery of genetic variations and development of genetic markers will facilitate research on rice genetics and genomics, and the exploration and utilization of elite gene resources in rice breeding. Among all genetic variations, single-nucleotide polymorphisms (SNPs) are the most abundant class of polymorphisms in the genome, and it is common for several SNPs to occur within a few hundred base pairs, which can enable a distinction to be made between highly similar cultivars (Ayed et al., Reference Ayed, Kallel, Hassen and Rebai2014; Gurgul et al., Reference Gurgul, Semik, Pawlina, Szmatoła, Jasielczuk and Bugno-Poniewierska2014). Furthermore, compared with other common genetic markers, such as simple sequence repeats and insertion–deletion markers, SNP analysis can be automated in high-throughput assays without the need for DNA separation by size (Wang et al., Reference Wang, Li, Kwon and Yang2016). Therefore, SNPs have increasingly become the markers of choice for accurate genotype identification, diversity analysis, and genome-wide association studies in many species (Li et al., Reference Li, Guo, Wang, Liu and Zou2015; Shavrukov, Reference Shavrukov2016). However, information available on SNPs in DXWR remains limited. The main objectives of this study were (1) to use genome sequencing data and bioinformatics approaches to detect genome-wide SNPs in DXWR and (2) to validate the reliability of the detected SNPs by polymerase chain reaction (PCR)-based Sanger sequencing.
Experimental
In this study, we used data from our previous study regarding whole-genome sequencing, reads filtering, and mapping to the published rice (Nipponbare) reference genome (http://rice.plantbiology.msu.edu/) (Kawahara et al., Reference Kawahara, de la Bastide, Hamilton, Kanamori, McCombie, Ouyang, Schwartz, Tanaka, Wu, Zhou, Childs, Davidson, Lin, Quesada-Ocampo, Vaillancourt, Sakai, Lee, Kim, Numa, Itoh, Buell and Matsumoto2013). The sequencing data of DXWR are deposited in NCBI with the accession number SRA167397 (Zhang et al., Reference Zhang, Zhang, Cui, Luo, Zhou and Xie2015). SNP loci in the consensus sequence were detected by comparing with the reference sequence and then filtered using specific requirements (quality value >20 and the result supported by at least two reads) using SoapSNP (Chagné et al., Reference Chagné, Crowhurst, Troggio, Davey, Gilmore, Lawley, Vanderzande, Hellens, Kumar, Cestaro, Velasco, Main, Rees, Iezzoni, Mockler, Wilhelm, Van de Weg, Gardiner, Bassil and Peace2012). To compare the detected SNPs with annotated gene structures in the rice genome, physical positions of DXWR sequences were integrated into the annotated RAP2 Nipponbare full-length cDNA database (Ohyanagi et al., Reference Ohyanagi, Tanaka, Sakai, Shigemoto, Yamaguchi, Habara, Fujii, Antonio, Nagamura, Imanishi, Ikeo, Itoh, Gojobori and Sasaki2006) using Generic Genome Browser software (http://gmod.org/wiki/GBrowse), as described by Stein et al. (Reference Stein, Mungall, Shu, Caudy, Mangone, Day, Nickerson, Stajich, Harris, Arva and Lewis2002).
Genomic DNA was extracted using the CTAB method (Porebski et al., Reference Porebski, Bailey and Baum1997). Based on the Nipponbare reference genome sequence, PCR primers specific for the sequences flanking the SNPs were designed. The details of the primers were presented in online Supplementary Table S1. PCR amplification was performed using the PrimeSTAR® Max DNA Polymerase Kit (Takara, Dalian, China). Target DNA fragments were cut from the agarose gel and purified using a DNA Gel Extraction Kit (Tiangen, Beijing, China), and then were sequenced at Shanghai Invitrogen Biotech (Shanghai, China). Sequence data were aligned using the publicly available programs Clustal X (http://www.clustal.org/) and BioEdit (http://www.mbio.ncsu.edu/bioedit/bioedit.html).
Discussion
In our previous study, whole-genome sequencing of DXWR was performed and 282,383,842 paired-end reads (90 bp per read) were obtained after filtering, and approximately 228 million reads were uniquely aligned with the reference genome of Nipponbare. An overall effective depth of 55× coverage was achieved, and the resulting consensus sequence was 357,407,193 bp, with 95.76% coverage of the Nipponbare genome (Zhang et al., Reference Zhang, Zhang, Cui, Luo, Zhou and Xie2015).
In this study, 1,089,478 SNPs were detected by genomic comparison between DXWR and Nipponbare (Table 1 and online Supplementary Table S2). The genome-wide average SNP density was one SNP per 0.33 kb, whereas the density varied among the chromosomes, from one SNP per 0.28 kb (chromosome 10) to one SNP per 0.41 kb (chromosome 8) (Table 1). Previously, Feltus et al. (Reference Feltus, Wan, Schulze, Estill, Jiang and Paterson2004) reported a genome-wide average SNP density of one SNP per 0.93 kb between 9311 (O. sativa ssp. indica) and Nipponbare, and Arai-Kichise et al. (Reference Arai-Kichise, Shiwa, Nagasaki, Ebana, Yoshikawa, Yano and Wakasa2011) reported a genome-wide average SNP density of one SNP per 2.9 kb between Omachi (O. sativa ssp. japonica) and Nipponbare. The high density of SNPs between DXWR and Nipponbare may reflect high sequence divergence among the diverse combinations used in our study. Furthermore, 166,903 SNPs were found to occur in the coding regions of 39,283 genes. Among these SNPs, 63,338 SNPs in 25,863 genes were synonymous, whereas 103,565 SNPs in 32,797 genes were non-synonymous, and 19,377 genes exhibited both synonymous and non-synonymous changes. The non-synonymous amino acid substitutions might be responsible for a change in the functional activity of the gene products.
Table 1. Densities of SNPs on individual chromosomes detected between DXWR and Nipponbare genomes
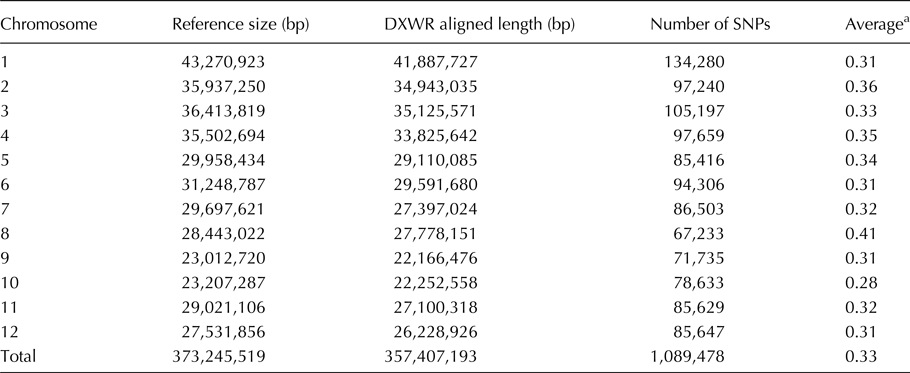
a The ‘Average’ column is a SNP density in DXWR, and equals to aligned length (bp)/number of SNPs/1000.
To assess the accuracy of SNPs detection by high-throughput sequencing, we randomly selected five SNPs to conduct PCR amplification and Sanger sequencing. A total of 10 PCR products were successfully amplified and sequenced between DXWR and the cultivated rice Nipponbare (online Supplementary Table S3). The 10 SNPs detected by high-throughput sequencing were all confirmed by PCR-based Sanger sequencing analysis (Fig. 1 and online Supplementary Table S3), indicating a high accuracy of SNPs detection in this study.
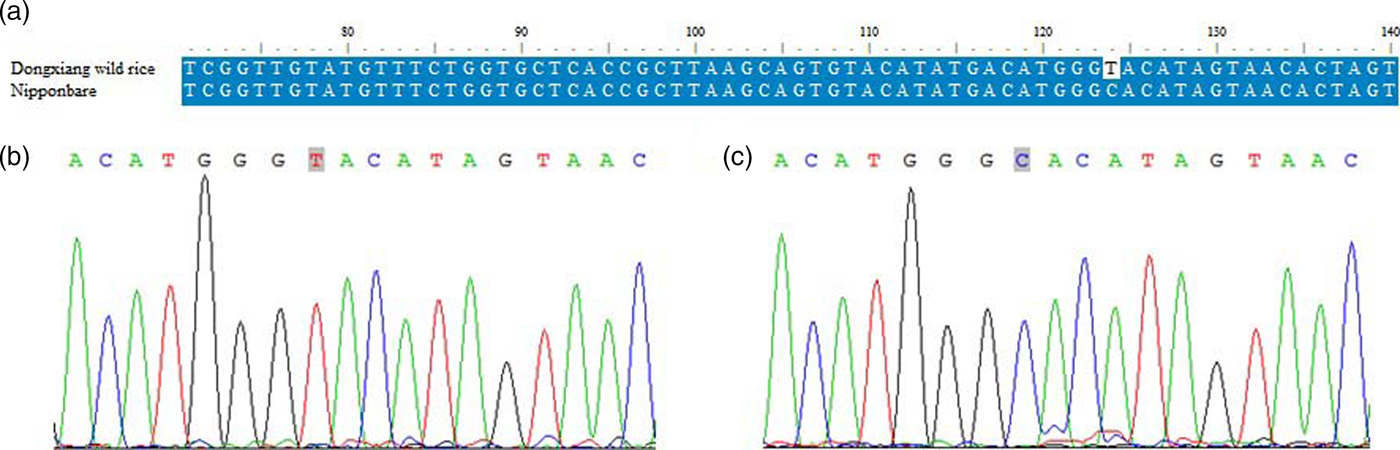
Fig. 1. Validation of the SNP locus (chromosome 5/19727118) and genotyping between DXWR and the cultivated rice Nipponbare by PCR-based Sanger sequencing. (a) SNP locus and its flanking sequences obtained from Sanger sequencing were aligned between DXWR and the cultivated rice Nipponbare. (b) Confirmation of the genotype (T) of the SNP locus in DXWR by Sanger sequencing. (c) Confirmation of the genotype (C) of the SNP locus in the cultivated rice Nipponbare by Sanger sequencing.
In conclusion, genome-wide SNPs were identified between DXWR and cultivated rice Nipponbare, and their accuracy was confirmed by PCR-based Sanger sequencing. The SNP markers identified in this study will provide a valuable resource for future research on genomics, evolution, genetic linkage, resource assessment, and genome-wide association in DXWR.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S1479262117000211.
Acknowledgements
This research was partially supported by the National Natural Science Foundation of China (31660386 and 31360327), the Key Projects of Jiangxi Education Department (KJLD12059), the Major Projects in Jiangxi Province (20161ACF60022), and the Sponsored Program for Distinguished Young Scholars in Jiangxi Normal University.




