INTRODUCTION
Toxoplasma gondii is a ubiquitous protozoan parasite that contributes to abortions and neonatal deaths amongst livestock as well as opportunistic infections in immunocompromised individuals, most notably AIDS patients (Dubey and Beattie, 1988). The ability to invade virtually all warm-blooded vertebrates most likely implies that T. gondii produces many antigens that play diverse roles in its invasion, replication and survival within the host cell. For instance, 3 different organelles, namely the micronemes, rhoptries and dense granules, secrete proteins that may contribute towards the parasite invasion process (Carruthers and Sibley, 1997). During the parasitic process, secretion of different proteins from these organelles takes place in sequence, eventually leading to parasite attachment and vacuole formation and modification (Carruthers and Sibley, 1997; Carruthers, 2002). The TgMIC2 microneme protein has been well-studied and found to be released from the apical complex of the parasite after binding to the host cell (Wan et al. 1997; Carruthers and Sibley, 1997). It has been proposed that TgMIC2 plays an important role in enabling the parasite to anchor itself onto the host cell to initiate the invasion process (Carruthers and Sibley, 1997; Carruthers, 2002). This is supported by the identification of specific adhesive motifs in the TgMIC2 DNA sequence belonging to the thrombospondin (TSP) family of adhesive proteins, which also include proteins such as thrombospondin-related anonymous protein (TRAP) and circumsporozoite protein-like thrombospondin-related protein (CTRP) of Plasmodium falciparum (Robson et al. 1988; Trottein, Triglia and Cowman, 1995) and Etp100 of Eimeria tenella (Tomley et al. 1991). Harper, Hoff and Carruthers (2004) have successfully shown that one of the adhesive motifs, the A-domain in TgMIC2, binds to heparin, a ubiquitous sulfated proteoglycan found in the extracellular matrix. Logically these adhesive motifs may serve as targets in blocking parasite attachment to healthy host cells.
In order to fully understand T. gondii infection and therefore design necessary therapeutic measures, the whole plethora of parasite proteins involved in the infection process should be characterized and eventually tested as potential vaccine candidates. Alternatively, antibodies against different key antigens of the parasite may be used as an antibody cocktail therapy approach (Elsaid et al. 1999; Prigione et al. 2000). Specific and high-affinity monoclonal antibodies are required as prophylactics against different parasite antigens. However, the production of monoclonal antibodies using the conventional hybridoma technology can take up to anywhere between 3 and 6 months, making it difficult to produce different monoclonal antibodies of diverse specificities in a short time. With the introduction of the phage display technology (Smith, 1985), monoclonal antibodies with high specificity and affinity can now be made within a month. This technology is based on linking antibody fragments to the bacteriophage coat protein. Each DNA fragment encoding a potential antibody is inserted into the phage genome thereby resulting in a physical fusion between the phage phenotype and genotype. Thus, affinity selection of antigen-specific antibodies can be performed directly from the collection of phage-bearing antibody clones and expression of the selected antibodies can be carried out in an Escherichia coli system.
In this study, we constructed a single-chain variable fragment (scFv) antibody phage display library from mice immunized with whole T. gondii. Screening the library against the recombinant TgMIC2 protein (rpTgMIC2) allowed us to isolate anti-TgMIC2 antibodies with good specificity to this antigen. This scFv library against total T. gondii parasite should allow us to isolate monoclonal antibodies against a multitude of different parasite proteins in a relatively short time.
MATERIALS AND METHODS
Expression and purification of rpTgMIC2
The construction and expression of rpTgMIC2 were performed according to Wan et al. (1997). The TgMIC2 gene was isolated through polymerase chain reaction (PCR) amplification using a T. gondii tachyzoite RH strain cDNA library (Wan, Blackwell and Ajioka, 1996) and cloned into the pET-22b(+) expression vector (Novagen, USA). Following transformation into the E. coli strain BL21, the isopropyl-β-D-thiogalactopyranoside (IPTG)-induced culture was centrifuged at 4000 g at 4 °C. The pellet was resuspended in 1× binding buffer without urea (imidazole 5 mM, NaCl 0·5 M, Tris-HCl 20 mM, pH 7·9) and sonicated (Vibracell, USA) for 30 sec at 50 Hz on ice. Following centrifugation at 10000 g for 15 min at 4 °C, the pellet was resuspended in 1× binding buffer with urea (imidazole 5 mM, NaCl 0·5 M, Tris-HCl 20 mM, urea 6 M, pH 7·9). The mixture was incubated overnight at 4 °C and centrifuged. The supernatant was loaded onto the metal affinity column (Novagen, USA) and washed with 1× binding buffer with urea, followed by 20 mM imidazole buffer (imidazole 20 mM, NaCl 0·5 M, Tris-HCl 20 mM, urea 6 M, pH 7·9) and eluted with 50 mM imidazole buffer (imidazole 50 mM, NaCl 0·5 M, Tris-HCl 20 mM, urea 6 M, pH 7·9).
Immunization of animals with T. gondii parasites
BALB/c mice were immunized with 106 T. gondii RH strain parasite particles using standard immunization procedures (Harlow and Lane, 1988). Prior to immunization, the parasites were suspended in phosphate-buffered saline (PBS) and killed in 2 freeze-thaw cycles and mixed with Freund's Incomplete Adjuvant (Sigma, USA). Two booster immunizations were given and at appropriate intervals, the mice were bled to produce serum samples whose titres were determined by indirect enzyme-linked immunosorbent assay (ELISA).
Wells of a 96-well microtitre plate were coated with 5 μg of parasite lysate or with 3% bovine serum albumin (BSA) in 50 μl of coating buffer (NaHCO3 0·1 M, pH 8·6) overnight at 4 °C. Wells were washed and blocked with 3% skim milk for 1 h at 37 °C. Diluted sera (1[ratio ]200 to 1[ratio ]25600) were added to the appropriate wells and incubated at 37 °C for 1 h. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1[ratio ]1000, Kirkegaard and Perry Laboratories, UK) and 2,2′-azinobis-(3-ethyl-benzothiazoline-6-sulphonic acid)-peroxide (ABTS-H2O2) substrate and peroxidase B solution (1[ratio ]1, Kirkegaard and Perry Laboratories, UK) were sequentially added into each well. Colour development was monitored at 405 nm. After demonstration of sufficient antibody titre, the mice were sacrificed and the spleens were harvested for total RNA isolation.
ScFv library construction
Total RNA was prepared from homogenized mice spleen using TRI-REAGENT (Molecular Research Center Inc., USA) according to the manufacturer's instructions. The resultant total RNA (20 μg) served as a template for cDNA synthesis (SUPERSCRIPT, Life Technologies, USA). The first strand cDNA was used as a template for amplification of the murine IgG variable heavy (VH) and variable light (VL) chain gene fragments. VH gene regions were amplified using a combination of 19 forward and 3 reverse primers respectively; while the V kappa gene regions were amplified using 17 forward and 3 reverse primers (Andris-Widhopf et al. 2001). V lambda gene regions were amplified using a set of forward and reverse primers only. For VH and V kappa gene amplifications, a total reaction volume of 100 μl containing 0·5 μg template cDNA, 60 pmoles of each forward and reverse primers, 1× PCR buffer, 2 mM MgCl2, 200 μM dNTP (Promega, USA), 2·5 units of Taq polymerase (Life Technologies, USA) was used. Forty amplification reactions were performed for each of the heavy and kappa light chain amplifications and 4 reactions were performed for the lambda light chain amplification. The amplifications were performed in the MJ Research PTC-100™ cycler (MJ Research, USA) with the following programme: 94 °C for 1 min, followed by 30 cycles of 94 °C for 15 sec, 56 °C for 30 sec and 72 °C for 90 sec, and concluded with a final elongation step at 72 °C for 10 min. The corresponding amplified products were purified using the Qiaquick Gel Extraction Kit (Qiagen, Germany). Subsequently, the purified light and heavy chain fragment coding sequences were fused in a second PCR using the primers RSC-F and RSC-B (Andris-Widhopf et al. 2001) to produce the single-chain variable antibody fragment (scFv). For this procedure, 50 reactions of 100 μl were prepared and each of these contained 100 ng of purified VH and VL genes respectively, 60 pmol of each RSC-F and RSC-B, 200 μM dNTP, 1× PCR buffer, 2·5 units of Taq polymerase (Life Technologies, USA) and sterile deionized water. The PCR reactions were performed with the following programme: 94 °C for 1 min, followed by 30 cycles of 94 °C for 15 sec, 56 °C for 30 sec and 72 °C for 2 min. ScFv fragments were gel purified and restricted with SfiI (20 U/μg DNA, 5 h at 50 °C). This mixture was subjected to gel purification and used in a test ligation as follows: 70 ng of DNA were ligated into 140 ng of SfiI restricted pComb3X vector (Scripps Research Institute, USA) (gel purified) with 1 U of ligase (Life Technologies, USA) in a 20 μl reaction, overnight at 15 °C. The ligated product was ethanol-precipitated and resuspended in 20 μl of sterile distilled water and transformed into electrocompetent ER2738 cells (New England Biolabs, USA) by electroporation (2·5 kV, 200 ohms, 25 μF). After the library size was estimated, 3 library ligations were performed by scaling up the reactions by a factor of 10 under similar conditions. The DNA was precipitated and transformed into ER2738 (F′ lacIq Δ(lacZ)M15 proA+B+ zzf::Tn10(TetR)/fhuA2 supE thi Δ(lac-proAB) Δ(hsdMS-mcrB)5 (rk−mk−McrBC)) cells.
Biopanning
The library was added to immobilized recombinant TgMIC2 protein (rpTgMIC2) (5 μg in 0·1 M NaHCO3, pH 8·6) and incubated for 2 h at 37 °C. Unbound phage were washed with 0·05× TBS-Tween and bound phage were eluted (glycine-HCl, pH 2·2), neutralized (1 M Tris-HCl, pH 9·1) and used to infect log phase ER2738 cells. Biopanning was repeated for a further 3 rounds with increasingly stringent washing (5 or 10 times) of unbound phage for each subsequent round. Phage selected from subsequent rounds of panning were amplified in the presence of the VCSM13 helper phage (Stratagene, USA). Input and output phage were titrated on LB-carbenicillin plates.
Phage ELISA
The original unpanned library and the resultant phage pool (output) from each subsequent round of panning were analysed by phage ELISA. For this, microtitre plate wells were coated with 5 μg of rpTgMIC2, and 50 μl of phage preparations were added and incubated for 2 h at 37 °C. Each well was extensively washed 10 times with sterile distilled water and HRP-conjugated anti-M13 secondary antibodies (Pharmacia, 1[ratio ]1000) were added, followed by an additional incubation of 1 h at 37 °C. Subsequently, ABTS-H2O2 substrate was added and colour development was monitored at 405 nm.
BstOI fingerprinting and sequencing
Output phage pools from the third and fourth rounds of panning were used to infect ER2738 cells. Following growth on LB-carbenicillin plates, 30 clones were selected from each round and phagemid DNA was extracted (RPM Kit, BIO 101). The scFv coding sequences in the plasmids were amplified using the ompseq and gback primer combination (Andris-Widhopf et al. 2001). The amplified products were digested with 15 U BstOI (Promega, USA) for 2 h at 60 °C, and fingerprints were analysed on 3% (w/v) agarose gels. ScFv inserts were sequenced by automated cycle sequencing using the ompseq primer for full-length sequence of the scFv, and HRLM-F primer (Andris-Widhopf et al. 2001) for the heavy chain variable region sequence.
Recombinant scFv expression and purification
To produce soluble scFv, phagemid DNA of selected clones were electroporated into TOP10 cells (Invitrogen, USA) (2·5 kV, 200 ohms, 25 μF) and following an overnight incubation at 37 °C on LB-carbenicillin plates, single colonies were inoculated into 20 ml SB (Super Broth). After a further incubation at 30 °C for 4–5 h (OD600 0·8–1·0), IPTG was added to a final concentration of 3 mM, followed by an overnight incubation at 30 °C. The cultures were centrifuged and the cell pellet was resuspended (Tris 5 mM, EDTA 1 mM, sucrose 20%) and subjected to 3 freeze-thawing cycles. The periplasmic products were stored at −20 °C. For purification purposes, the IPTG-induced cells were resuspended in 1× binding buffer without urea (imidazole 5 mM, NaCl 0·5 M, Tris-HCl 20 mM, pH 7·9). After 3 rounds of freeze-thawing, the cell lysate was centrifuged and the supernatant loaded onto a pre-equilibrated column with metal chelating resin (Novagen, USA) followed by washing with 1× binding buffer and buffer containing 50 mM imidazole. The scFv was eluted with 100 mM imidazole buffer and stored at −20 °C until required.
Western blotting
The presence of the scFv was detected by Western blotting (Towbin, Staehelin and Gordon, 1979). Expressed protein (periplasmic extract) was initially separated on a 14% (w/v) discontinuous SDS-polyacrylamide gel (Laemmli, 1970) and subsequently transferred to a nitrocellulose membrane. The membrane was blocked overnight in 5% (w/v) skim milk at 4 °C and followed by 3×5 min wash cycles with 0·1% (v/v) Tween 20 in PBS (PBST). The membrane was then incubated in peroxidase-conjugated mouse secondary anti-haemagglutinin (anti-HA) (1[ratio ]1000) and subsequently, ABTS-H2O2 substrate (1[ratio ]1) was added. The reaction was detected by exposing the membrane to X-ray film.
For the analysis of scFv specificity towards rpTgMIC2, 10 μg of rpTgMIC2 was separated on a 10% (w/v) discontinuous SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was blocked overnight (5% (w/v) milk in PBS), washed thoroughly and incubated with purified scFv for 2 h at room temperature, followed by 4×5 min washes with PBST. The membrane was then subjected to further treatments similar to that stated above.
RESULTS
Expression and purification of rpTgMIC2
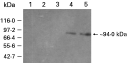
A protein band of ~94 kDa was observed in the crude extract of the induced rpTgMIC2 culture by SDS-PAGE, concomitant with the expected protein size of rpTgMIC2 (Wan et al. 1997) (data not shown). Following purification via the nickel column, only the 94 kDa protein was present, indicating that the rpTgMIC2 had been successfully purified from the other host cell protein contaminants.
Construction and biopanning of recombinant scFvs
cDNA was generated by reverse transcription from RNA isolated from spleens of mice immunized with whole cell T. gondii. Multiple reactions were performed for amplification of heavy and light chain variable fragments to avoid possible sequence bias in a single reaction. The success of a panning experiment largely depends on the diversity of the constructed combinatorial library and it is therefore important to increase the diversity of different V-genes amplified from the immune source. The VH forward primers have been designed to anneal to FR1 (framework-1) of the heavy chain while the VH reverse primers anneal to the FR4 region of the same antibody chain. Similarly, the forward and reverse primers for the amplification of the kappa and lambda light chains anneal to their respective FR1 and FR4 regions (Burton, 2001). The forward primers of the VL chain and reverse primers of the VH chain were designed to also incorporate SfiI restriction sites to facilitate subcloning into the SfiI-digested pComb3X phagemid vector system (Andris-Widhopf et al. 2001). In addition, the 3′ terminal of the amplified VL products and the 5′ terminal of the amplified VH products both have a short linker sequence that is responsible for the linkage of VL and VH products into a scFv antibody format (Fig. 1). The linker sequence contains 2 glycine and 4 serine residues that are commonly found in different linker sequences because glycine functions to enhance flexibility whilst serine increases the solubility of antibody molecules (Huston et al. 1988). These attributes and also the stability of the covalent bonding make the scFv a useful reagent in diverse applications.

Fig. 1. pComb3X-scFv construct. The scFv gene is inserted between the leader sequence ompA and gene III (gIII). Expression of scFv genes can be induced using IPTG and the scFv protein fragment will be fused to the histidine (His) and haemagglutinin (HA) peptide tags which are used respectively for scFv purification and identification purposes. The vector also carries an amber codon to allow the expression of soluble antibodies.
The amplified VH and VL products were electrophoresed and the expected fragment sizes of ~400 bp and ~350 bp respectively were observed (data not shown). The assembled scFv (~750 bp) was also obtained (data not shown) following overlap extension PCR. Recombinant phage, expressing a library of scFv genes on the phage pIII coat protein, were produced by VCSM13 helper phage rescue and biopanned against immobilized purified recombinant TgMIC2 protein. After the initial round of panning with the scFv library, the initial output number dropped in round 2 resulting in a low enrichment value of 0·02 due to the higher stringency in washing conditions (Table 1). However, after round 3, there was an increment in the number of specific phages as reflected in an enrichment value of 3-fold. This value steadily increased to 7·5-fold after the final panning round, indicating enhanced specific binding to rpTgMIC2. Phage ELISA was performed to detect the presence of enriched phage carrying scFv specific towards rpTgMIC2 over 4 rounds of biopanning. Phage pools from the original library and subsequent panning rounds were added individually to rpTgMIC2 coated and BSA coated (control) wells. The absorption value of the original phage pool was similar to the BSA control, plasmid (pComb3X) control and helper phage (VCSM13) control (Table 2). The value of the first round panned phage was 0·84-fold higher compared to the average value of the controls. In the second round, the value increased to 2·3-fold until a 6-fold increase was observed in the final panning round. The specificity of the interaction with rpTgMIC2 was evident by the lack of binding of the phage population to BSA.


Analysis of panned recombinant phages displaying scFv antibodies
The amplified selected phages were analysed by fingerprinting and sequencing of the antibody gene inserts. Thirty clones were each selected from the third and fourth panning rounds for further analysis. Phagemid DNAs were isolated from overnight cultures and the scFv gene inserts were amplified with the ompseq and gback primers. The ompseq primer anneals to the ompA signal peptide upstream of the SfiI restriction site (between 2620–2640 bp in pComb3X). The ompA directs transport of the protein to the inner membrane/periplasm of E. coli where the main pIII domain attaches the fusion protein to the tip of the assembling phage. In an amber suppressor host, this leader sequence will transport the recombinant antibody into the periplasm for modification to form active scFv protein. On the other hand, the gback primer is complementary to the C-terminal of gene III. The amplified product was about 750 bp and after digestion with BstOI and electrophoresis on 3% (w/v) agarose gels, the 60 clones were delineated into 2 groups based on the restriction profile (Fig. 2). Group 1 comprised of 38 clones whilst group 2 comprised of 22 clones. Two clones from each group were randomly chosen for automated DNA sequencing using two different primers and we observed that members within group 1 carried the same scFv sequence but the sequence differed from that of the members of group 2 (Fig. 3). The major diversity of the sequences from these different groups was contained within the complementarity-determining region 3 of the heavy chain (CDRH3), varying both in terms of sequence differences and length.

Fig. 2. Fingerprints of scFv clones obtained from panning against rpTgMIC2. ScFv genes were amplified from selected clones of round 3 (A) and round 4 (B), and digested with BstOI. Based on digestion profiles, clones were separated into group 1 (k1) and group 2 (k2). Lane M: 100 bp DNA marker (Promega, USA).

Fig. 3. Comparison of scFv sequences. Amino acid sequence alignment of representatives for scFv of group 1 (k1-3-rpTgMIC2) and group 2 (k2-9-rpTgMIC2), mouse IgG-kappa chain (GenBank Accession no: J04438) and mouse IgG-heavy chain (GenBank Accession no. M98041). CDRs and FRs represent the sequences for complementarity determining regions and framework regions respectively for VH and VL regions. Sequences were aligned with ClustalW (version 1.82) on the European Bioinformatics Institute (EBI) server <www.ebi.ac.uk/Tools/index.html> and displayed using Boxshade (version 3.21) <www.ch.embnet.org/software/BOX_form.html>. Identical residues are shaded dark and similar residues are shaded light. The linker sequence is in italics. Dashes (-) correspond to gaps inserted in the sequence to maximize the number of matched bases.
Recombinant scFv expression
The scFv gene was inserted into the pComb3X in a region downstream of the lacZ promoter and ompA, and upstream of the histidine and haemagglutinin peptide tags. ScFv expression was induced by IPTG and the resultant antibody fragments were fused to peptide tags of 6 histidine residues and haemagglutinin. The histidine residues are used for scFv purification purposes via nickel column chromatography whilst the haemagglutinin residues enable detection of the scFv antibodies during ELISA and Western blot experiments.
Two randomly-selected clones from each group were chosen for antibody expression and the scFvs were only detected for the group 1 (k1-3-rpTgMIC2; k1-2-rpTgMIC2) and not the group 2 (k2-9-rpTgMIC2; k2-1-rpTgMIC2) clones. The scFv antibody fragments from group 1 were observed to be ~32 kDa, whereby ~2 kDa of the total size was due to the presence of the histidine and haemagglutinin peptide tags (Fig. 4A). Some host proteins (~66 kDa) were also detected during the Western blotting due to the presence of 9 similar amino acid residues in certain E. coli proteins that can also be detected by the anti-haemagglutinin secondary antibody (Roche Molecular Chemicals, 1999). The expressed scFvs from clone k1-3-rpTgMIC2 were purified by metal affinity column chromatography whereby nickel ions were fixed to agarose resin and packed into a column. An SDS-PAGE gel profile and concomitant Western blot of the purified protein showed only 1 protein band with the expected size of ~32 kDa (Fig. 4B). Subsequently, the purified scFvs from clone k1-3-rpTgMIC2 were used in a Western blot experiment to determine the ability of binding to the panned protein (rpTgMIC2). Several controls were used and only the samples that contained the 94 kDa rpTgMIC2 exhibited a band on the autoradiogram (Fig. 5).

Fig. 4. Analysis of scFv expression and purification. (A) ScFvs expressed from group 1 clones, k1-3-rpTgMIC2 (lane 1) and k1-2-rpTgMIC2 (lane 2), and group 2 clones, k2-9-rpTgMIC2 (lane 3) and k2-1-rpTgMIC2 (lane 4), obtained from rpTgMIC2 biopanning, were probed with anti-HA monoclonal antibody. (B) Crude (lane 1) and purified (lane 2) scFvs expressed from the kl-3-rpTgMIC2 clone were analysed by SDS-PAGE (i) and Western blotting (ii). Protein sizes indicated were determined from broad range protein marker (New England Biolabs, USA).

Fig. 5. Detection of rpTgMIC2 using scFv from k1-3-rpTgMIC2. Lane 1: Host cell extract (E. coli BL21 (DE3) pLysS) without induction; Lane 2: host cell extract with induction (IPTG 1 mM final concentration); Lane 3: rc050-1 extract without induction; Lane 4: rc050-1 extract with induction; Lane 5: purified rpTgMIC2. rc050-1 is the clone that contains the recombinant expression plasmid for TgMIC2 gene. Protein sizes indicated were determined from broad range protein marker (New England Biolabs, USA).
DISCUSSION
We have constructed a phage-displayed scFv antibody library (1·02×106 cfu/μg DNA) against T. gondii RH strain of parasites. Four rounds of biopanning against rpTgMIC2 were performed leading to an enrichment value of 7·5-fold for phage displaying scFvs specific for rpTgMIC2. Commonly, an enrichment value of at least 50-fold indicates the success of the panning procedure to obtain specific binders (Barbas et al. 1991). A previous report on the isolation of scFvs against surface proteins of the gastrointestinal nematode parasite Haemonchus contortus utilized a similar approach as reported here but isolated scFvs enriched up to 300-fold (White, Meeusen and Newton, 2001). Therefore, with a lower-fold of enrichment, an excess of weak affinity scFvs have remained and caused outgrowth of low-affinity clones during the subsequent library amplification step. For a non-antigen specific library, a reduced number of specific and high-affinity binder clones in the initial library is a possibility. Nevertheless, although the phage output was reduced in round 2, the phage output increased after the third round indicating that binder clones had begun to be isolated and amplified successfully. The observation of amplification of rpTgMIC2 binders was also evident from the phage ELISA whereby as the stringency of the selection conditions increased, more phage displaying scFvs with better binding capabilities were successfully isolated and the increased proportion of these binder phages was reflected in the significant increase in absorbance (O.D.405nm) i.e. 0·05 in round 1 to 0·288 in round 3. It is also possible that the rpTgMIC2 does not completely mimic the structural conformation of native MIC2 protein secreted by T. gondii and this could have contributed to the low enrichment values as well as the lower specificity of the scFvs to total parasite crude extract (data not shown).
Thirty clones were randomly selected from rounds 3 and 4 for fingerprinting. The BstOI restriction enzyme was used because its restriction sites in the variable antibody gene region are random in position as well as in frequency. Thus the antibody clones can be delineated into different groups based on dissimilarities in size and number of DNA fragments after digestion with BstOI. When the sequences of the two different groups were compared to the database of mouse immunoglobulin genes, each sequence matched the characteristics of antibody gene sequences according to the Kabat definition of hypervariable (CDR) and framework (FR) regions (Kabat et al. 1991). Sequence analysis also indicated that the scFvs isolated in this study are not affinity matured. Nevertheless, they might be of value as an immunological tool, particularly for the analysis of the immune response against T. gondii. Distinct antigenic regions within the MIC2 antigen are recognized by T cells from both adults with acquired infection and children with congenital infection (Beghetto et al. 2005). Therefore, anti-MIC2 antibodies could be used to inhibit this adhesion molecule's role in parasite motility and invasion. Both the sequences of k1-3-rpTgMIC2 and k2-9-rpTgMIC2 were compared against each other and results showed a high level of dissimilarity typically in the CDRH3 regions which were highly variable in terms of sequence and length. This implies that both these scFvs probably recognized different epitopes of the same antigen. In addition, 67% of the antibody clones analysed after round 4 carried the same sequence as k1-3-rpTgMIC2 thereby indicating that this particular clone might have a higher affinity towards rpTgMIC2.
The pComb3X vector bearing the amber stop codon allowed for the expression of scFvs that were not fused to any additional phage protein residues (Barbas et al. 1991). Initially, the scFv antibody fragment was expressed in a format that was linked to the pIII phage coat protein due to the utilization of ER2738, a suppressor strain of the E. coli bacteria. In suppressor bacterial strains, expression of the amber stop codon is suppressed and therefore the scFv protein fragment was co-expressed with the amino terminal of the pIII coat protein, thus, allowing us to screen the library by biopanning. However, to enable expression of the free form of scFvs, phagemids from the selected clones were transformed into the E. coli TOP10 non-suppressor cell for expression studies. Only the representative group 1 clones expressed the recombinant scFvs, as was evident in the Western blot. Sequence checks on the representative group 2 clones did not reveal any introduction of mutational stops or abnormalities in the open reading frame of the antibody gene. Many factors can affect the efficiency of expression e.g. choice of nutrients and environmental parameters such as temperature, dissolved oxygen tension etc. The expression of foreign proteins in a recombinant host cell often utilizes significant amounts of host cell resources, placing a metabolic burden on the host, leading to a decrease in growth and a reduction of protein expression. Thus, several types of growth conditions and expression were employed such as the addition of glycerol, optimization of IPTG induction concentrations and reduction of induction time, but none of these optimizations led to the expression of the group 2 scFvs. Glycerol was used as an alternative carbon source (0·2% w/v) (Donovan, Robinson and Glick, 2000) whilst the induction time was reduced to prevent the possibility of scFv degradation due to inherent molecular instability (Studier et al. 1990). In addition, it is possible that different antibodies are expressed at different levels even when using the pComb3X vector expression system (Elia et al. 2001) and this problem may be overcome by subcloning of the scFv gene into a different expression vector.
The purified scFv from clone k1-3-rpTgMIC2 was shown to specifically recognize the rpTgMIC2, both in the crude extract as well as purified form. We had also attempted to demonstrate the scFv's ability to recognize and bind to native MIC2 protein in T. gondii crude extract. The scFv hybridized to a protein of 115 kDa which was also recognized by the anti-MIC2 monoclonal antibody, mAb6D10 (data not shown). Nevertheless, the scFv also recognized proteins of a lower molecular weight within the parasite extract indicating that scFvs of low specificity had been isolated during the 3 rounds of biopanning.
It is important to establish the specificity and affinity of the scFv to enable its use in therapeutic and diagnostic applications. Major improvements in DNA and phage display technology that address the problems faced when less specific scFvs are obtained include CDR mutagenesis (Chowdhury and Pastan, 1999). Mutations can also be introduced by way of error-prone PCR (Hawkins et al. 1993) and the use of bacterial mutator strains (Low, Holliger and Winter, 1996). The affinity of an antibody for its target antigen can also be improved upon by increasing the avidity of the monovalent scFv. By using shorter linkers, the monomeric scFv could combine with other scFv molecules to form diabodies, tribodies or even tetramers (LeGall et al. 1999) which would effectively increase the surface areas of the scFv that could bind to the antigen. A bispecific scFv constructed from k1-3-rpTgMIC2 and k2-9-rpTgMIC2 linked by a flexible peptide linker (Gly(4)-Ser)(3)) could possibly increase the selective MIC2-targeting properties.
We have presented proof of concept that a recombinant phage-displayed antibody library constructed from a whole cell immunized animal can be used to screen for antigen-specific binding antibodies. Nevertheless, the specificity of the antibodies should be improved upon by more stringent biopanning procedures or by re-panning the selected k1-3-rpTgMIC2 antibodies against properly folded rpTgMIC2 protein. A specific scFv antibody may have potential in therapy because it can be coupled with molecules such as drugs (Arap, Pasqualini and Rouslahti, 1998), toxins (Bera et al. 1998) or enzymes (Shabat et al. 2001), and has the ability to deliver these reagents accurately to the target sites. ScFvs that are specific to unknown T. gondii proteins can be coupled to such molecules and used to test their efficacy towards reducing or blocking parasite invasion into host cells. In instances where a T. gondii protein or antigen has an unknown location within the cell, the antigen-specific immunogold-labelled antibody fragment can be used to pinpoint the location of the protein by in situ hybridization or transmission electron microscopy (Carzaniga et al. 2002). This approach would also allow investigators to determine the role or localization of a particular T. gondii protein during the active stage of disease and also within the chronic cyst form. Similarly, the scFvs can be utilized for affinity purification of the corresponding native or recombinant antigen (Oh et al. 2003) for vaccine development. The development of vaccines against T. gondii is important and by the approach used in this study, it might be possible to specify relevant antigens or specifically, epitopes, which might be useful for vaccine development for stage specific toxoplasmosis. The approach presented here complements the whole genome phage display approach of Robben et al. (2002) that successfully identified an epitope of GRA3, an antigen located in the dense granules of T. gondii tachyzoites. In conclusion, antibody phage display technology is a promising approach in the study of antigen-antibody interactions for further elucidation of the pathogenesis of toxoplasmosis.
CONCLUSION
We have demonstrated that biopanning of a Toxoplasma gondii whole cell specific antibody phage display library could isolate monoclonal antibodies that bound to the specific antigen, rpTgMIC2. This library can serve as a pool of monoclonal antibodies to obtain high-affinity antibodies against many different T. gondii antigens. The availability of a plethora of monoclonal antibodies towards T. gondii proteins will allow further characterization of both antigens and antibodies. The antibodies can then be used as a tool to localize these antigens within whole T. gondii and hopefully increasing our understanding of this parasite and its pathogenesis.
This study was supported by the IRPA grant (09-02-02-0055) awarded by the Ministry of Science, Technology and the Environment, Malaysia and The Wellcome Trust Research Development Award in Tropical Medicine to K.L.W. L.N.H. is a recipient of the Malaysian National Science Fellowship. The authors are grateful to Dr J. Ajioka for his critical comments on the manuscript and to Dr D. Sibley for providing the mAb6D10 anti-MIC2 monoclonal antibody.