


Introduction
Chronic stress can precipitate depression in susceptible individuals, but the underlying molecular pathogenesis in the brain remains elusive. Similarly, antidepressants provide important relief in many individuals, but their mechanisms of action are not completely understood. Stress is used as a model to study alterations in brain structure and function because mood disorders are often precipitated or exacerbated by acute or chronic stressful life events (Reference Brown, Cooper-Kuhn and Kempermann1). Chronic unpredictable stress (CUS), consisting of several relatively unpredictable stressors, was considered to be a valid animal model of depression (Reference Willner2,Reference Willner3) and could induce anhedonia, a core symptom of depression in human, as measured by changes in sucrose consumption in rats (Reference Willner, Towell, Sampson, Sophokleous and Muscat4). The model was proved to be successful in the functional identification of antidepressant, and therefore had a high degree of predictive validity (Reference Muscat, Papp and Willner5–Reference Bondi, Rodriguez, Gould, Frazer and Morilak7).
There is increasing evidence suggesting cellular and molecular adaptations at several levels of brain neurons in response to antidepressant treatment. It has been reported that antidepressant treatment blocks the stress-induced atrophy of CA3 pyramidal cells and increases neurogenesis of hippocampal granule cells (Reference Duman8–Reference Duman and Monteggia10). Strong evidence suggests that antidepressants work by induction of neuroplastic changes mediated through regulation of brain-derived neurotrophic factor (BDNF). BDNF is a member of the neurotrophin superfamily that is responsible for promoting and modifying growth, development and survival of neuronal populations (Reference Lindsay, Wiegand, Altar and DiStefano11). The expression of BDNF in the hippocampus is dramatically downregulated by exposure to stress (Reference Smith, Makino, Kvetnansky and Post12). This effect is seen in the dentate gyrus (DG), CA3 and CA1 pyramidal cell layers, and is observed after acute or chronic stress. In contrast to the effects of stress, chronic administration of different classes of antidepressants increases the expression of BDNF in the hippocampus, as well as in the frontal cortex (Reference Duman8–Reference Duman and Monteggia10). Chronic administration of amitriptyline or venlafaxine at 5 mg/kg, but not 10 mg/kg, increased the intensity of BDNF immunostaining in hippocampal pyramidal neurons of normal rats (Reference Xu, Steven Richardson and Li13). Researchers have also found that a single infusion of BDNF into the hippocampus produces a potent and long-lasting antidepressant effect in these behavioural models (Reference Shirayama, Chen, Nakagawa, Russell and Duman14).
The transcription factor, cyclic-AMP response element binding protein (CREB), plays an important role in neuronal plasticity. CREB is dynamically phosphorylated at the serine 133 residue in response to a variety of neuronal stimuli. Therefore, nuclear staining of pCREB reports activation of a particular brain area in response to external or internal stimuli (Reference Shaywitz and Greenberg15). Increased expression of CREB would expect to lead to regulation of specific target genes, two of which may be BDNF and tyrosine kinase receptor B (TrkB) (Reference Nibuya, Morinobu and Duman16,Reference Nibuya, Nestler and Duman17). In addition, pCREB was associated with newly generated cells in the adult hippocampus and has been suggested to play a role in the formation of synaptic contacts by newly generated neurons in both the developing and adult brain (Reference Nakagawa, Kim and Lee18).
Venlafaxine is a dual-action antidepressant with high rates of remission achievement in major depression (Reference Rudolph19). The present study, in the adult rat hippocampus, was to evaluate the dose effects of chronic venlafaxine on the expression of BDNF and pCREB and the dose-related differences in the expression of neuroprotective proteins of rats.
Materials and methods
Animals
Adult male Sprague-Dawley rats weighing about 180–220 g were offered by the National Rodent Laboratory Animal Resources, Shanghai branch of China. One hundred rats were used in all experiments. Rats were housed in groups of eight in a temperature- and humidity-controlled (temperature: 21–23 °C; humidity: 50–55 °C) environment and maintained on a 12-h light/dark cycle with free access to food and water. All experiments were performed in accordance with the local, international and institutional guidelines. Every effort was taken to minimise the number of animals used and their suffering. Sixteen rats died during the course of the study.
Sucrose test
Sucrose test was used to measure anhedonia, which has been defined as a reduction in sucrose consumption relative to the control group. The protocol for this test was based on that reported previously (Reference Willner, Muscat and Papp20). Rats were first trained to consume 1% sucrose solution before beginning the experimental procedures. Training consisted of five 1-h baseline tests in which sucrose was presented in the home cage, followed by 24-h food and water deprivation; intake was measured by weighing the preweighted bottles containing the sucrose solution at the end of the test. Subsequently, sucrose consumption was monitored once each week, under similar conditions throughout the whole experiment. All sucrose tests were carried out at 8 a.m.
Open-field test (OFT)
The open-field test (OFT) evaluated the general locomotor and exploratory behaviour of rats, and the experiments were performed as described earlier (Reference Kennett, Dickinson and Curzon21). Each rat was placed at the centre of the open field (75 cm square chamber, 80-cm-high walls with its floor divided into 25 equal squares) for 5 min in a quiet room after weighing. Assessed parameters were the time in the centre square, the number of crossing squares, the frequency of rearing, the frequency of grooming and the number of fecal pellets. Next test was performed after cleaning the chamber. OFTs were performed before and after CUS.
CUS paradigm
The stressed rats were subjected to the following conditions used by Katz et al. (Reference Katz, Roth and Carroll22) and Willner et al. (Reference Willner, Muscat and Papp20), with minor modifications. Rats were subject to stress once per day over a period of 28 days between 8:00 and 12:00 a.m. The CUS procedure consisted of the following stressors in a random order: cold swim (4 °C, 5 min), food deprivation (48 h), hot swim (58 °C, 5 min), water and food deprivation (24 h), day and night reversion (24 h), tail pinch and shaker stress (2 min) and foot shock for 30 min (1 mA, 1 s duration, average 1 shock/min). Non-stressed animals were left undisturbed in the home cages except for the necessary controls such as regular cage cleaning and weighing.
Antidepressant treatment
After CUS, the stressed rats were divided into seven groups randomly. Normal control rats (NC, n = 10) and the first group of stressed rats (CUS0, n = 10) were killed (between 9 and 12 a.m.). The other stressed rats were treated for either 14 or 28 days with saline (14 days, CUS1, n = 10; 28 days, CUS2, n = 10) or venlafaxine [5 mg/kg (14 days, LV1, n = 11; 28 days, LV2, n = 11) and 10 mg/kg (14 days, HV1, n = 11; 28 days, HV2, n = 11)]. These doses have been widely used in animal studies (Reference Xu, Steven Richardson and Li13,Reference Gur, Dremencov, Lerer and Newman23). Venlafaxine was administered i.p. in a volume of 2 ml/kg with doses calculated from the salt form and expressed as milligrams per kilogram (Fig. 1).

Fig. 1. Details of the experimental procedure.
Immunohistochemistry
One day following the last dose of venlafaxine or saline, rats were anesthetised by i.p. injection of 10% chloral hydrate in normal saline. Rats were perfused transcardially with saline and then 4% paraformaldehyde in 0.1 M phosphate buffer (PB). After perfusion, the brains were extracted. Tissue blocks of 3-mm thick were cut and postfixed in the same fixative for 24 h.
Fixed blocks from the brain posterior to the infundibular stalk were embedded in paraffin. Coronal tissue sections of 4-µm thick were cut on a rotary microtome and mounted onto 3-amino-propyltriethoxy-silane (APES)-coated slides. Paraffin sections were dewaxed with xylene and rehydrated in a graded series of ethanol. Slides were submerged in 3% hydrogen peroxide to quench any endogenous peroxidase activity, washed with distilled water and heated at 95–98 °C in 1 mM ethylenediaminetetraacetic acid (EDTA) (pH 8.0) for 15 min, then cooled at room temperature for 40 min and washed with phosphate buffered saline (PBS). An aliquot of 10% non-immune goat serum was applied to eliminate non-specific staining. Primary antibodies were diluted in PBS and applied for 24 h at 4 °C (pCREB: 1:50, rabbit monoclonal, Cell signaling, USA; BDNF: 1:75, rabbit polyclonal, Chemicon, USA). The sections were washed with PBS and incubated with biotinylated goat anti-rabbit IgG antibodies for 30 min, rewashed with PBS and incubated with peroxidase-conjugated streptavidin for 30 min. The peroxidase activity was visualised by incubating the sections with a peroxidase substrate solution after sufficient washing. The sections were counterstained with haematoxylin and mounted. A 1:1000 diluted solution of non-immune rabbit serum was used as control in the immunohistochemical localisation of the pCREB and BDNF protein.
In order to determine pCREB and BDNF immunoreactivity intensity of hippocampus, sections were examined under bright-field illumination, homogeneously lighted and digitalised using a charge coupled device (CCD) camera. Photographs were taken at ×20 magnification. We counted the numbers and integral optical density (IOD) of positive cells.
Western blot
Right hippocampus of rats was rinsed with ice-cold PBS before being collected in lysis buffer. After lysis for 15 min in ice, the whole lysates were centrifuged at 13 400g for 15 min. The protein content in each supernatant fraction was determined using Bradford's solution, and samples containing an equivalent amount of protein were applied to 12% acrylamide denaturing gels (sodium dodecyl sulfate-polyacrylamide gel electrophoresis). After electrophoresis, proteins were transferred to nitrocellulose membranes (Amersham, Uppsala, Sweden) using a Bio-Rad mini-protein-III wet transfer unit overnight at 4 °C. Blotting membranes were incubated with 5% non-fat milk in Tris-buffered saline Tween-20 (TBST) [10 mM Tris (pH 7.6), 150 mM NaCl, 0.01% Tween-20] for 2 h at room temperature, washed three times, and then were incubated with phospho-CREB-ser133 (1:1000, rabbit monoclonal, Cell signaling, USA) and BDNF (1:1000, rabbit polyclonal, Chemicon, USA) in TBST overnight at 4 °C. After several washes with TBST buffer, the membranes were incubated for 1 h with horseradish peroxidase-linked secondary antibody (Santa Cruz, USA) diluted 1:10 000 followed by 10 washes. The membranes were then processed with enhanced chemiluminescence western blotting detection reagents (Pierce, Rockford, Illinois, USA). The films were scanned and densitometry was performed using the Labworks Version 4.5 image software (UVP, USA).
Reverse transcription polymerase chain reaction
Total RNA from the hippocampus was extracted using the Trizol reagent in accordance with the manufacturer's instructions. The RNA product was resuspended in 20 µl diethyl pyrocarbonate (DEPC)-treated water. The quality of RNA was judged from the pattern of ribosomal RNA after electrophoresis of RNA through 1.5% agarose gel containing ethidium bromide (EB) and visualisation by UV illumination. RNA was stored at −80 °C until use. After extraction of total RNA, reverse transcription was performed. Following incubation for 5 min at 72 °C, 2 µg of total RNA was reverse transcribed for 1 h at 42 °C with 0.01 µg random primers in the presence of 200 units of M-MLV reverse transcriptase (Promega, USA), 10 units of RNasin ribonulease inhibitor and 500 µM final concentration of deoxynucleotide triphosphate (dNTP) in a 20 µl reaction mixture. The cDNA products were stored at −20 °C until use.
The resulting single-stranded cDNA was amplified by polymerase chain reaction (PCR), which was performed with specific designed primers for the genes in search. After an initial 30 s denaturing cycle at 94 °C, an optimal number of cycles was performed as determined for the specific gene (Table 1), including denaturation, annealing and polymerisation, followed by a final 30 s elongation step at 72 °C. The optimal number of cycles and cDNA amount and primers' concentration were established according to a stringent calibration process determining the log–linear phase of amplification for each gene. The amplified products were separated on 1.5% agarose gels stained with EB and photographed under UV illumination with gel-documentation system. Results were evaluated as a relative unit and determined by normalisation of the IOD of CREB or BDNF band to that of the β-actin band.
Table 1 Prime sequences and PCR conditions

Statistical analysis
Results were presented as mean ± SD. Analysis of variance (ANOVA) for repeated measures was performed on sucrose intake and body weight. The results of open-field behaviour were analysed by one-factor ANOVA. When analysed all other measures, the data of NC and CUS0 were analysed by paired-sample t-test. The data of CUS1, CUS2, LV1, LV2, HV1 and HV2 were analysed by two-way ANOVA (time × dose). Where significant main effects or interactions were indicated, post hoc planned comparisons were performed using the Tukey test. Significance was established against an alpha level of 0.05.
Results
Change in body weight
There was a significant effect of stress treatment on the body weight of the rats. The mean body weight of rats in the eight experimental groups did not differ significantly initially. The weight gain in the groups exposed to CUS were significantly less than their controls at the end of the second, third and fourth week (Fig. 2).

Fig. 2. Effects of CUS and venlafaxine on body weight. NC, normal control group; CUS0, CUS group; CUS1, CUS + saline 14 days group; CUS2, CUS + saline 28 days group; LV1, CUS + venlafaxine (5 mg/kg) 14 days group; LV2, CUS + venlafaxine (5 mg/kg) 28 days group; HV1, CUS + venlafaxine (10 mg/kg) 14 days group; HV2, CUS + venlafaxine (10 mg/kg) 28 days group. n = 10–11 for each experimental group. Data were expressed as mean ± SD. The data were analysed of variance for repeated measures followed by Tukey post hoc test. *p < 0.05, NC group versus all the stressed group.
Measurement of sucrose intake
For the sucrose intake test, the last seven groups consumed sucrose solution much less than their controls at the end of the second, third and fourth week (Fig. 3).

Fig. 3. Effects of the CUS and venlafaxine on sucrose intake. NC, normal control group; CUS0, CUS group; CUS1, CUS + saline 14 days group; CUS2, CUS + saline 28 days group; LV1, CUS + venlafaxine (5 mg/kg) 14 days group; LV2, CUS + venlafaxine (5 mg/kg) 28 days group; HV1, CUS + venlafaxine (10 mg/kg) 14 days group; HV2, CUS + venlafaxine (10 mg/kg) 28 days group. n = 10–11 for each experimental group. Data were expressed as mean ± SD. The data were analysed of variance for repeated measures followed by Tukey post hoc test. *p < 0.05, NC group versus all the stressed groups.
Effect of CUS on open-field behaviour
Open-field testing is used to assess locomotion and exploration behaviour of rats or mice. The CUS paradigm increased the time in the centre square significantly. Compared with the controls, CUS decreased the frequency of rearing, the frequency of grooming and the number of crossing squares. No significant difference in the number of fecal pellets in the open tests was observed between CUS group and the control (Table 2).
Table 2 Effects of CUS on open-field behaviour during the 4-week study period
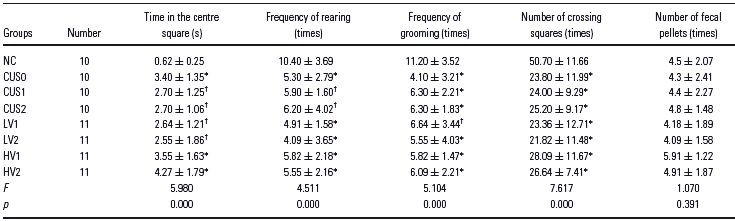
NC, normal control group; CUS0, CUS group; CUS1, CUS + saline 14 days group; CUS2, CUS + saline 28 days group; LV1, CUS + venlafaxine (5 mg/kg) 14 days group; LV2, CUS + venlafaxine (5 mg/kg) 28 days group; HV1, CUS + venlafaxine (10 mg/kg) 14 days group; HV2, CUS + venlafaxine (10 mg/kg) 28 days group. Data were expressed as mean ± SD. The data were analysed by one-factor ANOVA followed by Tukey post hoc test. Compared with control.
* p < 0.01.
† p < 0.05.
Immunohistochemistry
Rats subjected to CUS exhibited a specific inhibition of the number of pCREB and BDNF positive nuclei and IOD in the DG. There were no significant differences in the CA regions of the hippocampus between NC and CUS groups. Chronic low dose of venlafaxine (5 mg/kg) treatment for 28 days increased the number of pCREB positive nuclei and IOD in the DG, compared with CUS2 rats. Chronic administration of low dose venlafaxine (5 mg/kg, 14 and 28 days), but not high dose (10 mg/kg, 14 and 28 days), resulted in a significant increase in the BDNF IOD in the DG, compared to CUS1 and CUS2 rats. Two-way ANOVA revealed no time × dose interaction on the expressions of pCREB or BDNF protein (Fig. 4).

Fig. 4. Effects of CUS and venlafaxine on phosphorylated cAMP-responsive element binding protein (pCREB) (a) and brain-derived neurotrophic factor (BDNF) (b) immunoreactivity in the granular cell layer. Phase-contrast micrograph (×200) of NC, normal control group; CUS0, CUS group; CUS1, CUS + saline 14 days group; CUS2, CUS + saline 28 days group; LV1, CUS + venlafaxine (5 mg/kg) 14 days group; LV2, CUS + venlafaxine (5 mg/kg) 28 days group; HV1, CUS + venlafaxine (10 mg/kg) 14 days group; HV2, CUS + venlafaxine (10 mg/kg) 28 days group. Data were expressed as mean ± SD. n = 5 for each experimental group. The data were analysed by two-way ANOVA followed by Tukey post hoc test and paired-sample t-test. Scale bars: 100 µm.
Western blot
Rats subjected to CUS exhibited a specific inhibition of protein levels of pCREB and BDNF in the hippocampus. Chronic low dose of venlafaxine (5 mg/kg) treatment for 28 days increased the protein levels of pCREB compared with CUS2 rats. Chronic administration of low dose venlafaxine (5 mg/kg, 14 and 28 days), but not high dose, resulted in a significant increase in the protein levels of BDNF in the hippocampus, compared with CUS1 and CUS2. Two-way ANOVA revealed no time × dose interaction on the expressions of pCREB or BDNF protein (Fig. 5).

Fig. 5. Effects of CUS and venlafaxine on phosphorylated cAMP-responsive element binding protein (pCREB) and brain-derived neurotrophic factor (BDNF) in the hippocampus of the rat. (a) Western blot for pCREB; (b) western blot for BDNF; (c) quantitation of pCREB and BDNF between NC and CUS0; (d) quantitation of pCREB of CUS1, CUS2, LV1, LV2, HV1 and HV2 groups; (e) quantitation of BDNF of CUS1, CUS2, LV1, LV2, HV1 and HV2 groups. NC, normal control group; CUS0, CUS group; CUS1, CUS + saline 14 days group; CUS2, CUS + saline 28 days group; LV1, CUS + venlafaxine (5 mg/kg) 14 days group; LV2, CUS + venlafaxine (5 mg/kg) 28 days group; HV1, CUS + venlafaxine (10 mg/kg) 14 days group; HV2, CUS + venlafaxine (10 mg/kg) 28 days group. Data were expressed as mean ± SD. n = 5for each experimental group. The data were analysed by two-way ANOVA followed by Tukey post hoc test and paired-sample t-test.
Reverse transcription polymerase chain reaction (RT-PCR)
Rats subjected to CUS exhibited a specific inhibition of mRNA levels of CREB and BDNF in the hippocampus. Chronic administration of low dose venlafaxine (5 mg/kg, 14 and 28 days), but not high dose, resulted in a significant increase in mRNA levels of CREB in the hippocampus, compared with CUS1 and CUS2. There was a tendency for increased mRNA levels of BDNF in the LV1 and LV2 groups. Two-way ANOVA revealed no time × dose interaction on the expressions of CREB or BDNF mRNA (Fig. 6).

Fig. 6. The expression of β-actin, cAMP-responsive element binding protein (CREB) and brain-derived neurotrophic factor (BDNF)in the hippocampus after CUS and venlafaxine treatment. (a) RT-PCR for β-actin, CREB and BDNF; (b) quantitation of pCREB and BDNF between NC and CUS0; (c) quantitation of pCREB of CUS1, CUS2, LV1, LV2, HV1 and HV2 groups; (d) quantitation of BDNF of CUS1, CUS2, LV1, LV2, HV1 and HV2 groups. NC, normal control group; CUS0, CUS group; CUS1, CUS + saline 14 days group; CUS2, CUS + saline 28 days group; LV1, CUS + venlafaxine (5 mg/kg) 14 days group; LV2, CUS + venlafaxine (5 mg/kg) 28 days group; HV1, CUS + venlafaxine (10 mg/kg) 14 days group; HV2, CUS + venlafaxine (10 mg/kg) 28 days group. Data were expressed as mean ± SD. n = 5 for each experimental group. The data were analysed by two-way ANOVA followed by Tukey post hoc test and paired-sample t-test.
Discussion
It is generally believed that the CUS model is a widely used model of depression, which has induced several physiological and behavioural depressive-like symptoms (Reference Willner24). Several studies suggest that CUS-induced depression model can be used for evaluating the efficacy of antidepressant candidates through behavioural tests, such as sucrose preference and OFTs (Reference Willner, Towell, Sampson, Sophokleous and Muscat4,Reference Muscat, Papp and Willner5,Reference Mao, Xian, Ip, Tsai and Che25,Reference Larsen, Mikkelsen, Hay-Schmidt and Sandi26).
Sucrose preference test is an indicator of anhedonia-like behavioural change. Anhedonia, a core symptom of human major depression, was modelled by inducing a decrease in responsiveness to rewards reflected by a reduced consumption and/or preference of sweetened solutions. The results of present study showed that rats subjected to CUS procedure consumed less sucrose solution when compared with non-stressed rats. The CUS influenced exploratory activity, resulting in a reduction of the motility counts in our present study. This observation is consistent with an earlier study performed by Katz et al. (Reference Katz, Roth and Carroll22), involving exposure to a stress regime of which the CUS model is a development (Reference Willner, Muscat and Papp20).
Recent research considers that depression is associated with neuronal atrophy and dendritic reorganisation in the hippocampus and prefrontal cortex, and these changes are at least partially reversible by antidepressant treatment (Reference Radley, Sisti and Hao27–Reference Sheline, Wang, Gado, Csernansky and Vannier29). However, their mechanisms of action are not completely understood. In the current study, CUS significantly reduced hippocampal pCREB expression, and chronic low dose of venlafaxine treatment increased pCREB expression in the hippocampus. These are in line with the previous results (Reference Laifenfeld, Karry, Grauer, Klein and Ben-Shachar30,Reference Sairanen, O'Leary, Knuuttila and Castren31). CREB is important in the processes of axonal growth, as well as being involved in synaptic plasticity and processes of learning and long-term memory (Reference Davis, Vanhoutte, Pages, Caboche and Laroche32–Reference Viola, Furman and Izquierdo35). An increase in the antidepressant-induced pCREB, in contrast to the reduction in pCREB in the stress paradigm, supports the aforementioned implication of neuronal plasticity in stress and in the aetiology of depression and its treatment.
Currently, there are numerous studies suggesting that stress decreases the proliferation of new neurons in the subgranular zone (SGZ) of the hippocampus. One of the several different classes of antidepressants, including 5-hydroxytryptamine (5-HT) or NE-selective reuptake inhibitors, increases neurogenesis in adult hippocampus (Reference Warner-Schmidt and Duman36). Neurogenesis is a process of generating functionally integrated neurons from progenitor cells. In most mammals, active neurogenesis occurs throughout life in the subventricular zone of the lateral ventricles and in the SGZ of the DG (Reference Xu, Chen and He37). In the present study, pCREB immunoreactivity has changed most prominently in SGZ of the hippocampus after chronic stress and chronic venlafaxine treatment. Other investigators have previously reported that chronic antidepressant treatment increased pCREB expression in the hippocampus and DG of rats and humans (Reference Laifenfeld, Karry, Grauer, Klein and Ben-Shachar30,Reference Thome, Sakai and Shin38). Previous studies have showed that poly-sialated neural cell adhesion molecule (PSA-NCAM) was transiently expressed in recently generated granule cells and in neurons undergoing remodelling and plasticity (Reference Cremer, Chazal and Lledo39). Similarly, pCREB is expressed in immature neurons of the SGZ and is often colocalised with PSA-NCAM in these cells. Recently, our study has shown that CUS decreased the expression of NCAM mRNA in the hippocampus. Moreover, the expression of NCAM mRNA has been shown to increase after chronic low dose venlafaxine treatment (Reference Li, Yuan, Hou and Zhang40). Since chronic antidepressant treatment increases neurogenesis, and both PSA-NCAM and pCREB are markers of recently generated neurons, antidepressant-induced increases in neurogenesis may at least partially explain increased expression of PSA-NCAM and pCREB in the hippocampus of rats chronically treated with imipramine (Reference Sairanen, Lucas, Ernfors, Castren and Castren41). Taken together, our findings suggest that pCREB may play an important role in stress and in the treatment of depression.
In the current study, CUS significantly reduced hippocampal BDNF expression. Chronic low dose venlafaxine treatment (28 days) increased BDNF protein in the hippocampus. They are consistent with the previous studies (Reference Xu, Steven Richardson and Li13,Reference Nibuya, Nestler and Duman17). Initial studies reported significant reductions of BDNF mRNA expression in the DG granule, the CA3 and CA1 cell layer of the hippocampus after an acute immobilisation stress (Reference Smith, Makino, Kvetnansky and Post12,Reference Nibuya, Morinobu and Duman16). Our study showed that there was a trend towards increased mRNA levels of BDNF in the DG of chronic low dose venlafaxine treatment groups. Possibly, the reason for this discrepancy is due to differences in the experimental condition. Subsequent work found that other types of stress, including unpredictable, footshock, social isolation, social defeat, swim stress and maternal deprivation, also decreased the expression of BDNF in the hippocampus (Reference Duman and Monteggia10). In contrast to the actions of stress, different classes of antidepressants significantly increased the expression of BDNF in the major subfields of the hippocampus, including the granule cell layer and the CA1 and CA3 pyramidal cell layers (Reference Nibuya, Morinobu and Duman16,Reference Nibuya, Nestler and Duman17). The upregulation of BDNF by antidepressant treatment has been confirmed by a number of studies from different laboratories. However, there have been some inconsistent reports with certain classes of antidepressants. There are some studies that have not observed this effect (Reference Altar, Whitehead, Chen, Wortwein and Madsen42,Reference Dias, Banerjee, Duman and Vaidya43). Our study found that chronic low dose venlafaxine treatment increased the BDNF expression in the DG, but it did not induce a statistically significant BDNF expression in the CA1 and CA3 pyramidal cell layers. This could be due to the treatment paradigm, including the dose of drug or time of treatment (Reference Coppell, Pei and Zetterstrom44,Reference De Foubert, Carney and Robinson45). For example, once daily injections for 2 or 4 weeks were used in this study, while twice daily injections for 2 weeks or once daily injections for 3 weeks can also be used. We could also find that most drugs were given in similar or different doses.
Our results also suggest that changes in the pCREB and BDNF are related to the dose of venlafaxine being given. Consistently, a previous study reported that the high dose of venlafaxine decreased the intensity of BDNF immunostaining in all subareas of the normal rat hippocampus (Reference Xu, Steven Richardson and Li13). The increase in the pCREB and BDNF immunostaining in the hippocampus after the low dose of venlafaxine was not seen after the high dose. The high dose of venlafaxine had no significant effect on two plasticity-associated proteins in the CUS rat hippocampus. The mechanism responsible for their therapeutic action is not understood. The present results suggest that chronic venlafaxine treatment with low dose might be a more appropriate therapeutic method in CUS animal model.
The cAMP–CREB cascade is one of the important signalling pathways involved in antidepressant therapeutic mechanism, of which several molecular mechanisms had been found to be upregulated in the hippocampus after chronic antidepressant treatment, including cAMP-dependent protein kinase and the pCREB (Reference Nibuya, Nestler and Duman17,Reference Thome, Sakai and Shin38). Increased expression of CREB would in turn lead to the change of specific target genes, such as BDNF and TrkB. In the present study, the mRNA and protein of pCREB and BDNF were induced in the hippocampus almost at the same time point following venlafaxine administration, indicating that cAMP–CREB–BDNF cascade may play a significant role both in stress-related depression and in antidepressant effect.
Conclusion
These findings suggest that neuronal plasticity-associated proteins such as pCREB and BDNF play an important role both in stress and in the treatment of depression.
Acknowledgements
This study was partly supported by Nature Science Foundation of JiangSu Province (No: BK2009050, Yuan Yong-Gui), Key Program of Medical Development of Nanjing (No: ZKX07018, Yuan Yong-Gui), Nanjing Science and Technology Bureau Project Fund (QYK09185, Li Jing-Jing) and Nanjing Medical College Technological Fund (08NMUM117, Li Jing-Jing).








