1. Introduction
A breakdown in the capacity of cells to produce and distribute functionally folded proteins and to dispose of misfolded proteins has been implicated in many diseases. This dysfunction, often spurred by mutations, causes a variety of biochemical outcomes, including enhanced protein degradation, retention of proteins within the secretory pathway (Wiseman et al. Reference Wiseman, Koulov, Powers, Kelly and Balch2007a), formation of intracellular protein aggregates (Kopito, Reference Kopito2000), and deposition of amyloid fibrils in tissues (Selkoe, Reference Selkoe2003). These biological outcomes provide clues about the cellular components involved in pathogenesis and the biochemical nature of the dysfunction. However, the mechanisms by which these mutations trigger pathogenesis are often not obvious (Kelly & Balch, Reference Kelly and Balch2006; Powers et al. Reference Powers, Morimoto, Dillin, Kelly and Balch2009). This is especially true for mutations occurring in α-helical membrane proteins, which must fold, assemble, and maintain functional structures within the chemically diverse membranes of the endoplasmic reticulum (ER), the Golgi complex, the plasma membrane (PM), and other organelles (Kelly & Balch, Reference Kelly and Balch2006; Sanders & Mittendorf, Reference Sanders and Mittendorf2011). It is clear that the production of many wild-type membrane proteins in the cell is marginally efficient, which suggests that the energetics governing competing assembly and misassembly pathways is often comparable (Sanders & Nagy, Reference Sanders and Nagy2000). This may account for the fact that, in many cases, a multitude of diverse mutations is capable of prompting pathogenic misfolding of α-helical membrane proteins. For example, there are ca. 2000 mutations in the cystic fibrosis transmembrane regulator (CFTR) chloride channel that are known to cause cystic fibrosis, a majority of which are likely to induce misfolding as the primary cause of channel loss of function (cystic fibrosis mutation database: www.genet.sickkids.on.ca/cftr).
The precise origins of the cellular misfolding of α-helical membrane proteins remain elusive for several reasons. First, much less is known about the structure and conformational stability of α-helical membrane proteins compared to soluble proteins (White, Reference White2009). Indeed, technical limitations have long impeded the investigation of the structure and folding of membrane proteins in their native membrane solvent (Booth & Curnow, Reference Booth and Curnow2009; Stanley & Fleming, Reference Stanley and Fleming2008). Second, the synthesis, folding, and assembly of membrane proteins are elaborate cellular processes (Fig. 1a), which could potentially be disrupted in any one of a variety of ways (Fig. 1b–d) (Ng et al. Reference Ng, Poulsen and Deber2012). For these reasons, efforts to rationalize the mechanisms of the pathogenic misfolding of α-helical membrane proteins often encounter technical and conceptual challenges.

Fig. 1. Folding and misfolding of α-helical membrane proteins. Membrane protein biosynthesis involves several coupled processes, which are vulnerable to the influence of pathogenic mutations. (a) A cartoon depicts a typical biosynthetic pathway for an α-helical membrane protein. Biosynthesis begins with cotranslational integration of nascent α-helices (red) into the membrane (gray bar) by the translocon (gray donut), which is accompanied by early folding events (I). The nascent membrane protein (yellow) is released into the ER membrane following synthesis, where folding may continue with assistance of chaperones and folding enzymes (II). Once the protein achieves its native fold (green), it may form oligomeric complexes with potential interaction partners (blue) (III) prior to export from the ER. (b) Pathogenic mutations (red circle) may cause misincorporation of TM helices by the translocon, which establishes an incorrect topology for the nascent protein. (c) Pathogenic mutations may disfavor the formation of native tertiary or quaternary interactions. (d) Mutations may favor the formation of non-native contacts and/or aggregate formation. Here, we illustrate one of many possibilities, where the mutation both destabilizes the monomer structure and promotes formation of a non-native heterodimer.
Misfolding diseases are typically described as loss- or gain-of-function disorders; pathogenesis may arise as a result of the loss of functional protein due to misassembly or from the accumulation of cytotoxic protein aggregates (Cohen & Kelly, Reference Cohen and Kelly2003). The inherent linkage between events that lead to the loss of functional protein and those leading to the emergence of toxic protein aggregates allows related disease phenotypes to arise from distinct biochemical mechanisms. Indeed, characteristic phenotypes of misfolding diseases often stem from diverse genetic mechanisms, even within a single disorder. For example, the majority of cystic fibrosis (CF) patients carry the ΔF508 mutation in CFTR, which compromises its folding and biogenesis (Welsh & Smith, Reference Welsh and Smith1993). However, a number of other CFTR mutations are known to result in a loss of CFTR function through mechanisms other than misfolding (Ramsey et al. Reference Ramsey, Davies, Mcelvaney, Tullis, Bell, Drevinek, Griese, Mckone, Wainwright, Konstan, Moss, Ratjen, Sermet-Gaudelus, Rowe, Dong, Rodriguez, Yen, Ordonez, Elborn and Group2011; Sheppard et al. Reference Sheppard, Rich, Ostedgaard, Gregory, Smith and Welsh1993; Van Goor et al. Reference Van Goor, Hadida, Grootenhuis, Burton, Cao, Neuberger, Turnbull, Singh, Joubran, Hazlewood, Zhou, Mccartney, Arumugam, Decker, Yang, Young, Olson, Wine, Frizzell, Ashlock and Negulescu2009). Rapid identification of new disease-linked mutations resulting from the implementation of personal genome sequencing will provide new challenges and opportunities in mechanistic biology. Assessments of the effects of newly identified pathogenic mutations on the cellular processing, biochemical activity, folding, and structure of these proteins represents a laborious undertaking. Nevertheless, delineation of the effects of pathogenic mutations on α-helical membrane proteins may be critical for optimal design of therapeutics and, eventually, for the tailoring of therapeutic regimens. With regard to personalized medicine it should be emphasized that strategies to treat or avoid disease by correcting or avoiding misfolding of a protein are likely to be distinct from strategies to correct defects in other pathogenic variants of the very same protein with compromised function.
In this review, we discuss the physical principles governing the biogenesis and folding of α-helical membrane proteins and the potential influence of pathogenic mutations on these processes in the context of mammalian cells. Additionally, we highlight current progress and demonstrate potential applications of existing tools to rationalize the influence of pathogenic mutations on the biogenesis of α-helical membrane proteins.
2. Cotranslational folding and misfolding of α-helical membrane proteins
2.1. Translocon-mediated membrane integration of α-helical membrane proteins
In eukaryotic cells, translation of most α-helical membrane proteins occurs at the ER membrane and is mediated by the Sec61 translocon complex (Fig. 1a, step I). In addition to the Sec61 translocon itself, which consists of the integral membrane proteins Sec61 α, β, and γ (Denks et al. Reference Denks, Vogt, Sachelaru, Petriman, Kudva and Koch2014; Egea & Stroud, Reference Egea and Stroud2010; Van den Berg et al. Reference Van Den Berg, Clemons, Collinson, Modis, Hartmann, Harrison and Rapoport2004), the translocation process involves a number of accessory proteins that tune the function of the translocon complex and process the nascent chain (Johnson & van Waes, Reference Johnson and Van Waes1999; Schnell & Hebert, Reference Schnell and Hebert2003). During the early stages of membrane protein translation, the ribosome is docked to the translocon by the signal recognition particle (SRP), which effectively extends the ribosomal exit tunnel through the ER membrane and into the lumen. As translation continues at the ER membrane, the nascent polypeptide chain can access the ER membrane through the lateral gate of the translocon (Fig. 2; Heinrich et al. Reference Heinrich, Mothes, Brunner and Rapoport2000). Transmembrane (TM) helices pass through the lateral gate and into the ER membrane during translation, which establishes the initial topology of the α-helical membrane protein (topogenesis). Tertiary interactions between TM helices begin to form during translation (Cymer & von Heijne, Reference Cymer and Von Heijne2013; Khushoo et al. Reference Khushoo, Yang, Johnson and Skach2011; Meindl-Beinker et al. Reference Meindl-Beinker, Lundin, Nilsson, White and Von Heijne2006; Sadlish et al. Reference Sadlish, Pitonzo, Johnson and Skach2005), which represent the earliest steps of α-helical membrane protein folding (Fig. 1a, step I). The structural properties of cotranslational folding intermediates remain somewhat unclear. Thus, additional insights into the structure and function of the Sec61 translocon complex will ultimately be needed to enhance our understanding of the initial steps of α-helical membrane protein folding.
Fig. 2. Structure of the translocon. (a) A surface representation of the structure of the Pyrococus furiousus SecYEβ translocon in an open conformation (PDB code 3MP7) is shown. A yellow circle indicates the position of the protein conducting channel. The membrane is indicated in blue. (b) A cross-section of the structure reveals that the protein conducting pore is lined with both apolar and polar side chains. (c) A top down view depicts the opening of the lateral gate, through which nascent TM helices may access the ER membrane. (Figure from Öjemalm et al. Reference Öjemalm, Higuchi, Jiang, Langel, Nilsson, White, Suga and Von Heijne2011).
The structural and physical details of the events leading to the integration of individual nascent α-helices into the ER-membrane provide insights into the topogenic process. In many cases, the structure, dynamics, and functional mode of the translocon complex, which is regulated by a host of protein–protein interactions (Johnson & van Waes, Reference Johnson and Van Waes1999; Schnell & Hebert, Reference Schnell and Hebert2003; Snapp et al. Reference Snapp, Reinhart, Bogert, Lippincott-Schwartz and Hegde2004), may directly influence the manner by which individual helices are integrated into the membrane (Devaraneni et al. Reference Devaraneni, Conti, Matsumura, Yang, Johnson and Skach2011). Nevertheless, a sizeable body of knowledge on the translocon-mediated membrane integration mechanism has indicated that the selection of TM helices by the translocon is principally guided by the physiochemical properties of the nascent chain itself (White & von Heijne, Reference White and Von Heijne2008). Portions of the emerging polypeptide chain transiently sample both the hydrated interior pore of the translocon and a cross-section of the ER membrane in a manner that is well described by equilibrium partitioning models (Hessa et al. Reference Hessa, Kim, Bihlmaier, Lundin, Boekel, Andersson, Nilsson, White and Von Heijne2005; Öjemalm et al. Reference Öjemalm, Higuchi, Jiang, Langel, Nilsson, White, Suga and Von Heijne2011; White & von Heijne, Reference White and Von Heijne2008). Moreover, the elucidation of an empirical code for the energetics of translocon-mediated insertion has enabled reasonably accurate topogenic predictions from protein sequence (ΔG prediction server, www.dgpred.cbr.su.se) (Hessa et al. Reference Hessa, Kim, Bihlmaier, Lundin, Boekel, Andersson, Nilsson, White and Von Heijne2005; Kauko et al. Reference Kauko, Hedin, Thebaud, Cristobal, Elofsson and Von Heijne2010; Virkki et al. Reference Virkki, Agrawal, Edsbacker, Cristobal, Elofsson and Kauko2014). Translocon–bilayer partitioning energetics of amino acid side chains is generally consistent with both water–octanol and water–bilayer partitioning (Fleming, Reference Fleming2014; Moon & Fleming, Reference Moon and Fleming2011; White, Reference White2003; White & von Heijne, Reference White and Von Heijne2008; Wimley & White, Reference Wimley and White1996). Notably, the influence of a given amino acid on partitioning is strongly dependent on its position relative to the membrane (Hessa et al. Reference Hessa, Kim, Bihlmaier, Lundin, Boekel, Andersson, Nilsson, White and Von Heijne2005, Reference Hessa, Meindl-Beinker, Bernsel, Kim, Sato, Lerch-Bader, Nilsson, White and Von Heijne2007; Moon & Fleming, Reference Moon and Fleming2011), which reflects the position-dependent polarity of the bilayer solvent (White, Reference White2003; White & von Heijne, Reference White and Von Heijne2008). Partitioning of TM helices into the bilayer is dominated by the energetics associated with the burial of apolar surface area within the membrane core as well as by the positioning of positively charged residues among anionic phospholipid lipid head groups (positive-inside rule) (Öjemalm et al. Reference Öjemalm, Higuchi, Jiang, Langel, Nilsson, White, Suga and Von Heijne2011; von Heijne, Reference Von Heijne1986, Reference Von Heijne1992). These revelations provide a framework for understanding the sequence determinants of the early phase of α-helical membrane protein biogenesis and folding as well as the potential influence of pathogenic mutations on these processes.
2.2. Topogenesis of α-helical membrane proteins
The logic of the translocon suggests that a topogenic code should be written into the amino acid sequence of each α-helical membrane protein. Interestingly, a genomic survey of the predicted partitioning energetics of TM helices has revealed stark differences between single-pass and multi-pass α-helical membrane proteins (Hessa et al. Reference Hessa, Meindl-Beinker, Bernsel, Kim, Sato, Lerch-Bader, Nilsson, White and Von Heijne2007; White & von Heijne, Reference White and Von Heijne2008). In most cases, translocon-mediated insertion of the TM helices of single-pass α-helical membrane proteins appears to be highly favorable. This implies that membrane integration of single-pass TM helices is usually robust and may be insensitive to the influence of most single-point mutations. Nevertheless, there are likely to be some exceptions, as recent work by Feige and Hendershot has demonstrated that topogenesis of less hydrophobic single-pass TM helices can require the formation of complimentary interactions with the TM helices of its native interaction partners (Feige & Hendershot, Reference Feige and Hendershot2013).
Unlike single-pass membrane proteins, translocon-mediated membrane integration is predicted to be unfavorable for about one quarter of the TM helices in multi-pass α-helical membrane proteins (Hessa et al. Reference Hessa, Meindl-Beinker, Bernsel, Kim, Sato, Lerch-Bader, Nilsson, White and Von Heijne2007; White & von Heijne, Reference White and Von Heijne2008). These TM helices sometimes feature polar or charged residues positioned deep within the membrane, which are often critical for protein function or conformational stability (Adamian & Liang, Reference Adamian and Liang2002; Cao & Bowie, Reference Cao and Bowie2012; Gratkowski et al. Reference Gratkowski, Lear and Degrado2001; Illergard et al. Reference Illergard, Kauko and Elofsson2011; Popot & Engelman, Reference Popot and Engelman2000). The energetic penalty for burial of polar residues within the membrane may be partially offset by the formation of tertiary interactions between neighboring TM helices during translation (Meindl-Beinker et al. Reference Meindl-Beinker, Lundin, Nilsson, White and Von Heijne2006; White & von Heijne, Reference White and Von Heijne2008) or perhaps in some cases by the formation of transient hydrogen bonds with buried water molecules, which are often found in the crystal structures of α-helical membrane proteins (Miyano et al. Reference Miyano, Ago, Saino, Hori and Ida2010). However, pathogenic mutations that introduce non-native polar side chains within TM helices, which constitutes the most common class of amino acid substitution for disease-linked point mutations in α-helical membrane proteins (Partridge et al. Reference Partridge, Therien and Deber2004), bear the potential to disrupt these critical interactions. Both the influence of pathogenic mutations on topogenesis and the ramifications of cotranslational misfolding on cellular proteostasis merit further consideration.
The molecular details of multi-pass α-helical membrane protein topogenesis have been intensely studied for a number of proteins. In particular, the topogenesis of CFTR has served as a key model system (Kim & Skach, Reference Kim and Skach2012; Sadlish & Skach, Reference Sadlish and Skach2004). One interesting property revealed by these studies is the heterogeneous nature of its biosynthetic pathway. For instance, two charged residues within the first TM segment of CFTR prevent its efficient recognition as a TM helix during the early steps of biogenesis (Lu et al. Reference Lu, Xiong, Helm, Kimani, Bragin and Skach1998). Because the initial topology of the N-terminal TM domains influences that of the subsequently synthesized TM helices (Chen & Zhang, Reference Chen and Zhang1999; Kanki et al. Reference Kanki, Sakaguchi, Kitamura, Sato, Mihara and Hamasaki2002), inefficient recognition of the first TM helices may ultimately cause topological heterogeneity in the nascent structural ensemble. Characterization of the topology of various truncated CFTR constructs suggests that ca. 70% of the nascent proteins eventually acquire the correct topology in the first two TM helices (Lu et al. Reference Lu, Xiong, Helm, Kimani, Bragin and Skach1998; Xiong et al. Reference Xiong, Bragin, Widdicombe, Cohn and Skach1997). It is tempting to speculate that this topological heterogeneity may contribute to the poor efficiency with which even wild-type CFTR is known to correctly fold and traffic to the cell surface (Ward & Kopito, Reference Ward and Kopito1994). Topological heterogeneity appears to be a feature of the nascent forms of a number of multi-pass α-helical membrane proteins including P-glycoprotein (Pgp, MDR1) (Moss et al. Reference Moss, Helm, Lu, Bragin and Skach1998; Skach et al. Reference Skach, Calayag and Lingappa1993), sarcoplasmic/ER Calcium ATPase 2 (SERCA2) (Bayle et al. Reference Bayle, Weeks and Sachs1995), anion exchanger-1 (AE1, band 3) (Kanki et al. Reference Kanki, Sakaguchi, Kitamura, Sato, Mihara and Hamasaki2002), and aquaporin-1 (AQP1) (Buck & Skach, Reference Buck and Skach2005; Lu et al. Reference Lu, Turnbull, Bragin, Carveth, Verkman and Skach2000; Skach et al. Reference Skach, Shi, Calayag, Frigeri, Lingappa and Verkman1994). In some cases, the misincorporation of entire TM helices can occur during the biosynthesis of topologically ‘frustrated’ membrane proteins (Gafvelin & von Heijne, Reference Gafvelin and Von Heijne1994), which feature sequences with ambiguous topogenic codes (von Heijne, Reference Von Heijne2006). Studies of AE1 (Kanki et al. Reference Kanki, Sakaguchi, Kitamura, Sato, Mihara and Hamasaki2002), rhodopsin (Kanner et al. Reference Kanner, Klein, Friedlander and Simon2002), and AQP1 (Lu et al. Reference Lu, Turnbull, Bragin, Carveth, Verkman and Skach2000; Virkki et al. Reference Virkki, Agrawal, Edsbacker, Cristobal, Elofsson and Kauko2014) biosynthesis have shown that aberrant topomers of nascent proteins may be corrected post-translationally. However, to our knowledge, the mechanisms and molecular players involved in correcting aberrant topomers are currently unclear. Regardless of the mechanism, the reorientation of TM helices can sometimes require hours (Lu et al. Reference Lu, Turnbull, Bragin, Carveth, Verkman and Skach2000) and may often be outpaced by the rapid degradation of misassembled topological intermediates by the proteasome (Buck & Skach, Reference Buck and Skach2005).
Non-ideal topogenesis of multi-pass α-helical membrane proteins is consistent with the prediction that the translocon–bilayer partitioning equilibrium of many TM helices within these proteins is predicted to be close to 0 kcal mol−1 (Hessa et al. Reference Hessa, Meindl-Beinker, Bernsel, Kim, Sato, Lerch-Bader, Nilsson, White and Von Heijne2007; White & von Heijne, Reference White and Von Heijne2008), suggesting that only a fractional population of the nascent TM helices should spontaneously assume the correct topology during translation. These findings highlight the inherent plasticity of the translocon-mediated membrane integration process and again suggest the potential for facile distortion of this process by pathogenic mutations.
2.3. Influence of pathogenic mutations on the translocon-mediated insertion of TM helices
The apparent biochemical inefficiency of topogenesis suggests a potentially disruptive role for pathogenic mutations at the translocon. To our knowledge, it is not clear whether this is a common effect of pathogenic mutations. We utilized the ΔG prediction server to survey the effects of 470 non-synonymous mutations known to be associated with misfolding diseases occurring within or near the TM helices of five multi-pass α-helical membrane proteins including rhodopsin, vasopressin V2 receptor (V2R), CFTR, peripheral myelin protein 22 (PMP22), and the voltage-gated potassium channel KCNQ1 (manuscript in preparation). In the absence of tertiary contacts, 13 of the 36 total TM helices are predicted to insert with moderate efficiency (−1 kcal mol−1 < ΔG app < 1 kcal mol−1) and 7 are predicted to insert with poor efficiency (ΔG app > 1 kcal mol−1). Of the surveyed pathogenic mutations implicated in misfolding diseases, 63 mutations (ca. 10%) were predicted to increase the predicted free energy for the insertion of their respective TM helices by more than 1 kcal mol−1 (disfavoring insertion). Furthermore, 31 of these 63 mutations occur within TM helices predicted to have moderate or poor insertion efficiency in the wild-type protein. The true partitioning behavior of these TM helices and the magnitude of the energetic effects of these mutations are likely to be different in the context of the full-length protein due to the formation of helical hairpins within the translocon (Engelman & Steitz, Reference Engelman and Steitz1981; Heinrich & Rapoport, Reference Heinrich and Rapoport2003; Hermansson & von Heijne, Reference Hermansson and Von Heijne2003; Meindl-Beinker et al. Reference Meindl-Beinker, Lundin, Nilsson, White and Von Heijne2006). Nevertheless, it seems feasible that some of these pathogenic mutations could interfere with topogenesis. Furthermore, the rapid degradation of aberrant topological intermediates (Buck & Skach, Reference Buck and Skach2005) suggests a mechanism by which pathogenic mutations that influence topogenic efficiency may decrease the yield of mature protein. The considerations outlined in this section suggest that the induction of misfolding by mutations that interfere with topogenesis are not rare, but may be much less common than mutations that disrupt or alter later stages of folding (discussed below). Testing this hypothesis is an avenue for future research.
3. Energetics of folding and misfolding of α-helical membrane proteins
3.1. Physical principles of post-translational α-helical membrane protein folding
As is true for soluble proteins (Anfinsen, Reference Anfinsen1973), the conformational trajectories of membrane proteins seek free-energy minima (Fleming, Reference Fleming2014; Huang et al. Reference Huang, Bayley, Liao, London and Khorana1981; Kim et al. Reference Kim, Schafer and Wolynes2014; Popot et al. Reference Popot, Trewhella and Engelman1986; Stanley & Fleming, Reference Stanley and Fleming2008; White, Reference White2003). Furthermore, the conformational energy landscapes of α-helical membrane proteins may dictate the nature of their post-translational interactions with the cellular quality control machinery (Roth & Balch, Reference Roth and Balch2011; Sanders & Myers, Reference Sanders and Myers2004). After TM helices are inserted into the bilayer by the translocon, the helices associate in order to establish their native tertiary structure (Fig. 1a, step II; White, Reference White2003); a process often rationalized by the two-stage model (Engelman et al. Reference Engelman, Chen, Chin, Curran, Dixon, Dupuy, Lee, Lehnert, Matthews, Reshetnyak, Senes and Popot2003; Popot & Engelman, Reference Popot and Engelman1990). Despite long-standing interest in this phenomenon, practical limitations have long hampered the characterization of the tertiary folding of α-helical membrane proteins under equilibrium conditions (Booth & Curnow, Reference Booth and Curnow2009; Hong et al. Reference Hong, Joh, Bowie and Tamm2009). As a result, our current understanding of the conformational energy landscapes of α-helical membrane proteins remains rudimentary (Bowie, Reference Bowie2005; Kim et al. Reference Kim, Schafer and Wolynes2014).
Surveying the energy landscapes of α-helical membrane proteins ideally involves measurement of the kinetic and thermodynamic barriers separating native and non-native states occurring within biological membranes; a daunting challenge. Nevertheless, the characterization of experimentally tractable conformational equilibria has revealed a number of fundamental principles. Despite the low dielectric environment within the membrane, the energetic contribution of hydrogen bonds to membrane protein conformational equilibria appears similar to that of soluble proteins (Bowie, Reference Bowie2011; Faham et al. Reference Faham, Yang, Bare, Yohannan, Whitelegge and Bowie2004; Joh et al. Reference Joh, Min, Faham, Whitelegge, Yang, Woods and Bowie2008; Li et al. Reference Li, You and Hristova2006). The energetic contribution of van der Waals packing interactions also appears to be similar for soluble proteins and α-helical membrane proteins (Doura et al. Reference Doura, Kobus, Dubrovsky, Hibbard and Fleming2004; Faham et al. Reference Faham, Yang, Bare, Yohannan, Whitelegge and Bowie2004; Fleming et al. Reference Fleming, Ackerman and Engelman1997; Joh et al. Reference Joh, Oberai, Yang, Whitelegge and Bowie2009). However, unlike soluble proteins, membrane proteins are subject to forces imposed by cellular membranes. Various lines of evidence involving both α-helical and β-barrel membrane protein folding in lipid bilayers have suggested that the width and curvature of the membrane can significantly influence folding reactions (Allen et al. Reference Allen, Curran, Templer, Meijberg and Booth2004a; Booth & Curnow, Reference Booth and Curnow2009; Brown, Reference Brown2012; Burgess et al. Reference Burgess, Dao, Stanley and Fleming2008; Hong & Tamm, Reference Hong and Tamm2004). Determination of the means by which these forces combine to shape the conformational equilibria of α-helical membrane proteins in membranes represents a frontier in protein science (Dill & MacCallum, Reference Dill and Maccallum2012).
3.2. Conformational energetics of multi-pass α-helical membrane proteins
Efforts to probe the basic features of the conformational energy landscapes of multi-pass α-helical membrane proteins have proven challenging due, in part, to the fact that commonly used denaturing agents such as urea are rarely capable of sufficiently disrupting their conformational equilibria (Fleming, Reference Fleming2014; Stanley & Fleming, Reference Stanley and Fleming2008). For this reason, such studies have relied heavily on the use of mild detergent micelles or detergent–lipid mixed micelles that energetically favor the native ensemble, which can be titrated with a charged denaturing detergent (i.e. SDS) that promotes the formation of a denatured ensemble. Perturbation of the conformational equilibrium can be accomplished in mixed micelle systems by modulating the mole fraction of the denaturing detergent. Providing that reversibility can be achieved (Fleming, Reference Fleming2014; Moon et al. Reference Moon, Kwon and Fleming2011), observations of the conformational ensemble upon the addition or dilution of the denaturing detergent can provide quantitative insights into the kinetic and thermodynamic properties of the conformational equilibrium. Such experiments facilitate the measurement of an equilibrium between the native ensemble and a non-native ensemble that typically retains some α-helical secondary structure but lacks tertiary or quaternary structure (Dutta et al. Reference Dutta, Kim, Moeller, Wu, Alexiev and Klein-Seetharaman2010; Krishnamani et al. Reference Krishnamani, Hegde, Langen and Lanyi2012; Lau & Bowie, Reference Lau and Bowie1997; London & Khorana, Reference London and Khorana1982; Riley et al. Reference Riley, Wallace, Flitsch and Booth1997; Schlebach et al. Reference Schlebach, Kim, Joh, Bowie and Park2011; Stanley & Fleming, Reference Stanley and Fleming2008). Although mixed micelle solvents are certainly not a perfect proxy for biological membranes (Matthews et al. Reference Matthews, Zoonens and Engelman2006; Warschawski et al. Reference Warschawski, Arnold, Beaugrand, Gravel, Chartrand and Marcotte2011; Zhou & Cross, Reference Zhou and Cross2013), the loss of tertiary structure and partial loss of secondary structure that accompanies denaturation of α-helical membrane proteins with a charged detergent is generally consistent with the documented structural defects in the misfolded forms of pathogenic rhodopsin variants responsible for retinitis pigmentosa (Liu et al. Reference Liu, Garriga and Khorana1996). Thus, assessment of the conformational equilibria of α-helical membrane proteins in mixed micelles may provide insight into the nature of the energetic transitions relevant to the misfolding process. Nevertheless, strategies to measure conformational stability of proteins within bilayers and even native membranes are on the horizon. For instance, the recent advent of the ‘steric trap’ approach by Hong and Bowie (Fig. 3), has enabled quantitative assessments of conformational equilibria under bilayered membrane conditions (Chang & Bowie, Reference Chang and Bowie2014; Hong et al. Reference Hong, Blois, Cao and Bowie2010; Hong & Bowie, Reference Hong and Bowie2011). Application of the steric trap has already demonstrated, quite strikingly, that dimerization of glycophorin A seems to be weaker in membranes than in micelles (Hong & Bowie, Reference Hong and Bowie2011). Thus, many aspects of the nature of the interplay between the conformational equilibrium of membrane proteins with biological membranes remain to be explored.
Fig. 3. Steric trap method originally developed by Heedeok Hong and James Bowie to assess the conformational equilibria of a membrane protein by measuring interactions between biotinylated membrane proteins and engineered monovalent streptavidins (mSA). Biotin labels are introduced at two proximal sites in the native conformation of the protein of interest (black circles). Binding of a single mSA to one of the biotins attached to the folded membrane protein occurs with high affinity. Binding of a second mSA to the other biotin can take place only after the membrane protein spontaneously unfolds. Unfolding and binding of this second mSA are therefore thermodynamically coupled. The position of the equilibrium between the single mSA-bound folded protein and the mSA-double bound unfolded form is assessed as a function of mSA concentration using any one of a variety of possible methods (unfolding-induced dissociation of a fluorescent excimer pair is implied in this example). Changes in the observed signal as a function of [mSA] can be fit for the equilibrium constant for unfolding (K unfold) by accounting for the known binding affinity of mSA for avidin. This method relies on use of an engineered series of mSA with K d,avidin spanning several orders of magnitude. Choice of the optimal engineered mSA to use is based on the need to tune the balance between the binding and unfolding equilibria.
The conformational stability of a handful of purified α-helical membrane proteins have been quantitatively assessed in various membrane mimetics. Interestingly, conformational stability measurements of diacylglycerol kinase (DAGK) (Lau & Bowie, Reference Lau and Bowie1997), bacteriorhodopsin (bR) (Curnow & Booth, Reference Curnow and Booth2007), the KcsA potassium channel (Barrera et al. Reference Barrera, Renart, Poveda, De Kruijff, Killian and González-Ros2008), and aquaglyceroporin GlpF (Veerappan et al. Reference Veerappan, Cymer, Klein and Schneider2011) have suggested large thermodynamic separation of the native and denatured reference states (ΔG unf = 16–31 kcal mol−1). On the other hand, studies of disulfide bond-reducing protein B (DsbB) (Otzen, Reference Otzen2003), galactose transporter GalP (Findlay et al. Reference Findlay, Rutherford, Henderson and Booth2010), lactose permease LacY (Harris et al. Reference Harris, Findlay, Simms, Liu and Booth2014), human PMP22 (Schlebach et al. Reference Schlebach, Peng, Kroncke, Mittendorf, Narayan, Carter and Sanders2013), and the rhomboid protease (Baker & Urban, Reference Baker and Urban2012) have suggested a more modest thermodynamic preference for their native conformations (ΔG unf = 0–4·5 kcal mol−1). Can it be that the range of thermodynamic stabilities of single-domain wild-type α-helical membrane proteins varies by more than an order of magnitude? This remains to be resolved. However, in addition to technical issues involving empirical stability extrapolations (Chang & Bowie, Reference Chang and Bowie2014; Schlebach et al. Reference Schlebach, Cao, Bowie and Park2012; Sehgal et al. Reference Sehgal, Mogensen and Otzen2005), the seemingly large dynamic range of stability measurements may arise, in part, from the distinct structural and energetic properties of the unfolded states generated by the different denaturants employed in these studies (Stanley & Fleming, Reference Stanley and Fleming2008). Indeed, a recent work by Chang and Bowie has revealed that the proposed free-energy difference between native bR and SDS-denatured bR is about twice as large as that separating native bR from a sterically trapped unfolded bR ensemble under identical phospholipid bicelle conditions in the absence of SDS (Chang & Bowie, Reference Chang and Bowie2014; Curnow & Booth, Reference Curnow and Booth2007). Comparisons of the thermodynamic stabilities is also complicated by the fact that unfolding is coupled to changes in the oligomeric state for some of these proteins (DAGK, KcsA, and GlpF), the energetics of which are dependent on the protein-to-detergent (and/or lipid) ratio utilized in the chosen reaction conditions (Fleming, Reference Fleming2002). Despite such difficulties, apples-to-apples comparisons of stability measurements for wild-type and mutant variants in mixed micelle systems have proven widely useful (Baker & Urban, Reference Baker and Urban2012; Cao & Bowie, Reference Cao and Bowie2012; Curnow & Booth, Reference Curnow and Booth2009; Curnow et al. Reference Curnow, Di Bartolo, Moreton, Ajoje, Saggese and Booth2011; Joh et al. Reference Joh, Oberai, Yang, Whitelegge and Bowie2009; Otzen, Reference Otzen2011). Nevertheless, the steric trap method (Chang & Bowie, Reference Chang and Bowie2014; Hong et al. Reference Hong, Blois, Cao and Bowie2010; Hong & Bowie, Reference Hong and Bowie2011), alternative applications of current methods (reviewed in (Hong et al. Reference Hong, Joh, Bowie and Tamm2009)), and possibly new approaches will be needed to characterize the thermodynamic preference for the native ensemble within bilayered vesicles and actual biological membranes.
Conformational energy landscapes also dictate the rates of α-helical membrane protein folding. The rates of soluble protein folding reactions vary greatly but are generally rapid (Plaxco et al. Reference Plaxco, Simons, Ruczinski and Baker2000); folding of soluble proteins generally requires anywhere from microseconds to minutes in vitro. Similar to soluble proteins (Brockwell & Radford, Reference Brockwell and Radford2007), kinetic intermediates of helical membrane proteins can form within milliseconds of the initiation of folding from detergent-denatured states (Allen et al. Reference Allen, Curran, Templer, Meijberg and Booth2004b; Booth et al. Reference Booth, Flitsch, Stern, Greenhalgh, Kim and Khorana1995; Krishnamani & Lanyi, Reference Krishnamani and Lanyi2011; Lu & Booth, Reference Lu and Booth2000; Otzen, Reference Otzen2003). However, complete refolding and/or oligomerization can require anywhere from minutes to days in vitro (Allen et al. Reference Allen, Curran, Templer, Meijberg and Booth2004b; Cao et al. Reference Cao, Schlebach, Park and Bowie2011; Jefferson et al. Reference Jefferson, Blois and Bowie2013; Krishnamani & Lanyi, Reference Krishnamani and Lanyi2011; Riley et al. Reference Riley, Wallace, Flitsch and Booth1997; Schlebach et al. Reference Schlebach, Cao, Bowie and Park2012, Reference Schlebach, Peng, Kroncke, Mittendorf, Narayan, Carter and Sanders2013). The folding of the soluble denatured forms of β-barrel membrane proteins into membranes also appears to be quite slow (Burgess et al. Reference Burgess, Dao, Stanley and Fleming2008; Gessmann et al. Reference Gessmann, Chung, Danoff, Plummer, Sandlin, Zaccai and Fleming2014; Huysmans et al. Reference Huysmans, Baldwin, Brockwell and Radford2010, Reference Huysmans, Radford, Baldwin and Brockwell2012), though in this case the rate-limiting step seems to involve the transfer of the unfolded protein from the aqueous phase to the membrane phase (Gessmann et al. Reference Gessmann, Chung, Danoff, Plummer, Sandlin, Zaccai and Fleming2014; Huysmans et al. Reference Huysmans, Baldwin, Brockwell and Radford2010, Reference Huysmans, Radford, Baldwin and Brockwell2012). Slow-folding relative to biological time scales suggest an essential biological role for chaperones and folding enzymes, a possibility consistent with the observed differences in the chaperone binding of wild-type CFTR relative to the slow-folding pathogenic ΔF508 variant (Coppinger et al. Reference Coppinger, Hutt, Razvi, Koulov, Pankow, Yates and Balch2012; Qu et al. Reference Qu, Strickland and Thomas1997; Wang et al. Reference Wang, Venable, Lapointe, Hutt, Koulov, Coppinger, Gurkan, Kellner, Matteson, Plutner, Riordan, Kelly, Yates and Balch2006). Thus, characterization of folding intermediates and their reactivity with the components of cellular quality control may be central to understanding the folding and misfolding mechanisms of pathogenic variants (Roth & Balch, Reference Roth and Balch2011).
The cellular turnover of α-helical membrane proteins is also likely to depend on their rates of unfolding (kinetic stability). For many soluble proteins (Park et al. Reference Park, Zhou, Gilmore and Marqusee2007; Xia et al. Reference Xia, Manning, Hesham, Lin, Bystroff and Colón2007), conformational stability is effectively achieved through a high kinetic barrier to unfolding (Jaswal et al. Reference Jaswal, Sohl, Davis and Agard2002). Several lines of evidence indicate that some α-helical membrane proteins may also be kinetically isolated from non-native states in some cases. For instance, the extrapolated unfolding rate of bR in mixed micelles suggests that accessing the detergent-denatured ensemble under native conditions would require a very, very long time (Curnow & Booth, Reference Curnow and Booth2007, Reference Curnow and Booth2010). Furthermore, the dissociation and/or unfolding of trimeric DAGK in lipid bicelles has been found to require weeks (Jefferson et al. Reference Jefferson, Blois and Bowie2013). High kinetic barriers to unfolding and/or dissociation of oligomers would be consistent with the slow subunit exchange observed for both DAGK and AcrB in membranes (Jefferson et al. Reference Jefferson, Blois and Bowie2013; Lu et al. Reference Lu, Chai, Zhong, Yu, Fang, Wang, Li, Zhu and Wei2012). Thus, in contrast to rapidly folding and unfolding single-domain soluble proteins, it is unclear whether most membrane proteins effectively achieve an equilibrium conformational ensemble in vivo. One intriguing possibility is that nascent membrane proteins freely interconvert between native and non-native conformations in the ER in the presence of the chaperones of quality control and low cholesterol concentrations, but become kinetically trapped in their native states after export to the Golgi and beyond.
Collective elucidation of the interplay between the conformational stability, folding rates, and unfolding rates of α-helical membrane proteins may ultimately be necessary to rationalize the cellular proteostasis of α-helical membrane proteins.
3.3. Influence of pathogenic mutations on α-helical membrane protein folding and assembly
Numerous diseases are linked to the defective folding and/or trafficking of membrane proteins. The cellular biosynthesis and assembly of membrane proteins is an inherently inefficient process (Ellgaard & Helenius, Reference Ellgaard and Helenius2003; Sanders & Nagy, Reference Sanders and Nagy2000) and pathogenic variants are typically assembled with even lower efficiencies, which suggests that these mutations cause structural defects that are detected by cellular quality control machinery (Kaushal & Khorana, Reference Kaushal and Khorana1994; Naef & Suter, Reference Naef and Suter1999; Sanders & Myers, Reference Sanders and Myers2004; Sung et al. Reference Sung, Schneider, Agarwal, Papermaster and Nathans1991). However, little is known about the influence of pathogenic mutations on the conformational equilibria of such proteins. This impasse primarily stems from the restricted number of α-helical membrane proteins that have thus far been amenable to in vitro investigations of folding and assembly. Nevertheless, a survey of the nature of pathogenic side-chain substitutions and their distributions throughout the topological domains of α-helical membrane proteins suggests that these mutations are likely to disrupt their conformational stability (Sanders & Myers, Reference Sanders and Myers2004). For example, the currently identified pathogenic mutations in PMP22 are distributed throughout its sequence, with many involving the introduction of charged or helix-breaking residues within its TM helices (Fig. 4). In the case of rhodopsin, a majority of pathogenic mutations seem to fall within a cluster of residues predicted to be essential for folding (Rader et al. Reference Rader, Anderson, Isin, Khorana, Bahar and Klein-Seetharaman2004). Regardless of their position within the protein or the nature of the substitution, a majority of tested membrane protein mutations have been seen to destabilize tertiary or quaternary structure (Baker & Urban, Reference Baker and Urban2012; Cao et al. Reference Cao, Schlebach, Park and Bowie2011; Curnow & Booth, Reference Curnow and Booth2009; Curnow et al. Reference Curnow, Di Bartolo, Moreton, Ajoje, Saggese and Booth2011; Faham et al. Reference Faham, Yang, Bare, Yohannan, Whitelegge and Bowie2004; Fleming & Engelman, Reference Fleming and Engelman2001; Nagy & Sanders, Reference Nagy and Sanders2004; Otzen, Reference Otzen2011), which may potentially explain why pathogenic mutations in α-helical membrane proteins are often well-distributed throughout the sequence. The magnitude of the effects of mutations on the conformational equilibria of α-helical membrane proteins seem to be on par with that of soluble proteins. Furthermore, a semi-quantitative investigation of DAGK folding has suggested that the degree of destabilization imparted by point mutations is inversely correlated with kinetic stability (Nagy & Sanders, Reference Nagy and Sanders2004), which suggests that an increase in the free energy of the native ensemble may be a common effect of such point mutations. Mutations that specifically destabilize the rate-limiting transition state for folding may be relatively rare, but have been documented (Curnow et al. Reference Curnow, Di Bartolo, Moreton, Ajoje, Saggese and Booth2011; Nagy & Sanders, Reference Nagy and Sanders2002; Otzen, Reference Otzen2011). Alternatively, a number of pathogenic mutations have been found to promote the formation of alternative structures via non-native contacts and/or disulfide bonds (Fig. 1d) (Dhaunchak & Nave, Reference Dhaunchak and Nave2007; Goldberg et al. Reference Goldberg, Loewen and Molday1998; Hwa et al. Reference Hwa, Reeves, Klein-Seetharaman, Davidson and Khorana1999; Li et al. Reference Li, You and Hristova2006; Ng & Deber, Reference Ng and Deber2010; Therien et al. Reference Therien, Grant and Deber2001; You et al. Reference You, Spangler, Li, Han, Ghosh and Hristova2007). Thus, it seems that cellular misfolding of α-helical membrane proteins may arise as a result of disruption of native contacts, formation of non-native contacts, or some combination of both.
Fig. 4. Topology of human PMP22 and distribution of pathogenic missense-encoded single amino acid mutations within its sequence. Residues for which pathogenic variants have been identified are indicated in red, and the identities of the pathogenic side-chain substitutions are indicated. Mutations in PMP22 result in Charcot–Marie–Tooth disease and related disorders.
New insights into the influence of pathogenic mutations on the tertiary folding of α-helical membrane proteins have been afforded by recent characterizations of the conformational stability and the structural dynamics of human PMP22, the cellular misfolding of which causes a spectrum of peripheral neuropathies (Jetten & Suter, Reference Jetten and Suter2000; Li et al. Reference Li, Parker, Martyn, Natarajan and Guo2012b; Sanders et al. Reference Sanders, Ismail-Beigi and Mcenery2001; Suter & Snipes, Reference Suter and Snipes1995). As of 2014, the Human Gene Mutation Database lists 44 point mutations in PMP22 that are known to cause Charcot–Marie–Tooth disease (CMTD), Dejerine–Sottas syndrome (DSS), or hereditary neuropathy with liability to pressure palsies (HNPP; hgmd.cf.ad.uk) (Fig. 4). The ΔG prediction server (www.dgpred.cbr.su.se) suggests favorable translocon-mediated membrane integration for each of the four TM helices of PMP22 and few of these mutations are predicted to significantly influence topogenesis (manuscript in preparation). Thus, the primary sequence suggests that the TM helices should spontaneously partition into the membrane even in the absence of favorable tertiary contacts. For this reason, it seems likely that pathogenic mutations in PMP22 primarily influence folding and assembly processes that occur after translocon-mediated membrane integration, which is consistent with the observation that the cellular trafficking of pathogenic PMP22 variants is stalled at the so-called ‘intermediate compartment’ between the ER and the Golgi (Tobler et al. Reference Tobler, Notterpek, Naef, Taylor, Suter and Shooter1999).
Emerging evidence suggests that the tertiary structure of wild-type PMP22 is only marginally stable and that most pathogenic mutations further reduce conformational stability. Reversible unfolding measurements of wild-type PMP22 have revealed a minimal free-energy difference between folded and unfolded conformations in n-dodecylphosphocholine (DPC) micelles (ΔG unf ≈ 0 kcal mol−1) (Schlebach et al. Reference Schlebach, Peng, Kroncke, Mittendorf, Narayan, Carter and Sanders2013; Fig. 5a). It is, of course, likely that PMP22 possesses a somewhat greater degree of conformational stability in biological membranes. Nevertheless, slow folding and marginal stability of wild-type PMP22 in cellular membranes could potentially account for its poor cellular trafficking efficiency (only ~20% of cellular WT PMP22 reaches the PM) and rapid cellular degradation (T 1/2 ≈ 30 min) (Pareek et al. Reference Pareek, Notterpek, Snipes, Naef, Sossin, Laliberté, Iacampo, Suter, Shooter and Murphy1997). Characterization of the effects of the quintessential pathogenic mutations L16P and G150D, which cause CMTD in mice and humans (Suter et al. Reference Suter, Moskow, Welcher, Snipes, Kosaras, Sidman, Buchberg and Shooter1992), has suggested that these mutations both decrease conformational stability and enhance the aggregation propensity of PMP22 (Myers et al. Reference Myers, Mobley and Sanders2008; Tobler et al. Reference Tobler, Liu, Mueller and Shooter2002). Furthermore, a comparison of the dynamics of WT and L16P PMP22 by NMR has revealed that this mutation enhances transient dissociation of the first TM helix and the transition of its other three TM segments into a molten globular domain (Fig. 5b) (Sakakura et al. Reference Sakakura, Hadziselimovic, Wang, Schey and Sanders2011), which may suggest that the nature of the unfolding reactions responsible for cellular misfolding of PMP22 may be relatively minor. We have recently expanded our analysis of pathogenic PMP22 variants and found that most pathogenic mutations do indeed destabilize its tertiary structure and decrease its cellular trafficking efficiency (manuscript in preparation). These findings will provide much needed insight as to how the conformational stability of pathogenic α-helical membrane proteins variants is tied to their cellular fates.
Fig. 5. Folding and conformational dynamics of human PMP22. (a) WT PMP22 was equilibrated in mixed micelles containing DPC in the absence (diamonds) and presence (circles) of 15% glycerol and varying levels of the denaturing detergent lauroyl sarcosine (LS). For each point tertiary structural content was monitored using the near-UV circular dichroism signal at 299 nm ([θ]299). Closed symbols are for a forward titration (unfolding) with LS. Open symbols are for a reverse (refolding) titration. The match between the forward and reverse titration data indicates the complete reversibility of the unfolding/refolding transitions, as confirmed by complementary kinetic studies (Schlebach et al. Reference Schlebach, Peng, Kroncke, Mittendorf, Narayan, Carter and Sanders2013). Kinetic and thermodynamic analyses suggest that the fraction of folded PMP22 in the absence of 15% glycerol (diamonds) is close to 0·5 in DPC micelles in the absence of LS (ΔG unf ≈ 0 kcal mol−1). PMP22 is stabilized in the presence of 15% glycerol, which shifts the unfolding transition to higher mole fractions of denaturing detergent under this condition (circles). However, a fit of the unfolding transition to a two-state equilibrium model (black line) suggests that the folded conformation is still only marginally favored in the presence of glycerol (ΔG unf = 1·5 ± 0·1 kcal mol−1). (Figure from Schlebach et al. Reference Schlebach, Peng, Kroncke, Mittendorf, Narayan, Carter and Sanders2013) (b) The structural dynamics of WT and L16P PMP22 were assessed in tetradecylphosphocholine (TDPC) micelles using solution NMR and other methods. The results indicate that the initial state of PMP22 unfolding involves dissociation of its first TM segment from the four-helix bundle, with the remaining three-helix bundle adopting a molten globule-like state in the membrane. The pathogenic L16P mutation breaks the first TM segment and shifts the folding equilibrium towards the partially unfolded state. (Modified figure from Sakakura et al. Reference Sakakura, Hadziselimovic, Wang, Schey and Sanders2011).
Although mutations in PMP22 appear to influence the native tertiary structure, many other pathogenic mutations are known to perturb the oligomerization of α-helical membrane proteins. Dimerization and activation of receptor tyrosine kinases (RTK) is a critical step in a number of cellular signaling pathways and mutations within the TM domains of these proteins are known to cause a various forms of cancer and developmental disorders. Investigation of pathogenic RTK variants in vitro has provided strong evidence that these mutations can promote unregulated RTK dimerization through the formation of non-native hydrogen bonds or perhaps non-native disulfide bonds (Li et al. Reference Li, You and Hristova2006; You et al. Reference You, Spangler, Li, Han, Ghosh and Hristova2007). Interestingly, quantitative studies of such mutations in vivo have suggested that energetic effects of mutations as little as 0·5 kcal mol−1 can have profound consequences on biochemical activity and cellular turnover under certain circumstances (Chen et al. Reference Chen, Degnin, Laederich, Horton and Hristova2011; He & Hristova, Reference He and Hristova2008; Placone & Hristova, Reference Placone and Hristova2012). Despite these advances, exhaustive efforts to identify consensus sequence motifs specifying the interaction of TM helices or to predict the effects of mutations on dimerization from structure have been somewhat unsuccessful (Li et al. Reference Li, Wimley and Hristova2012a; MacKenzie & Fleming, Reference Mackenzie and Fleming2008). Thus, the rationalization of the effects of pathogenic mutations on the folding and assembly of α-helical membrane proteins in the cell presently remains a pressing challenge in membrane protein biophysics.
4. Proteostasis and the cellular trafficking of α-helical membrane proteins
4.1. α-Helical membrane protein misfolding in the context of ER quality control
Both in concert with and following translocon-mediated integration of TM segments into the membrane of the ER, eukaryotic membrane proteins form tertiary and quaternary structures to attain their native structural state (Fig. 1a). During this process, membrane proteins interact with a host of proteins that are dedicated to faciliation of the folding process and also to policing of the misfolding of nascent proteins. These proteins include chaperones, prolyl isomerases, disulfide-bond isomerases, glycosyltransferases, glycosidases, ubiquitin ligases, and proteases. In this section, we explore the later stages of membrane protein folding and misfolding in the context of the complex environment of mammalian cells. Elucidation of the molecular basis of misfolding diseases will ultimately require interpretation of the effects of these conformational defects on the native (and non-native) interactions of the protein with other proteins as well as lipids and metabolites (Ellgaard & Helenius, Reference Ellgaard and Helenius2003; Kelly & Balch, Reference Kelly and Balch2006; Vembar & Brodsky, Reference Vembar and Brodsky2008). Elucidation of pathogenic mechanisms will also hinge on our understanding of the response of cells to dramatic changes in the flux of misfolded proteins caused by these mutations (Brodsky & Skach, Reference Brodsky and Skach2011).
In many cases, only a fraction of nascent membrane proteins achieve maturity (Pareek et al. Reference Pareek, Notterpek, Snipes, Naef, Sossin, Laliberté, Iacampo, Suter, Shooter and Murphy1997; Sanders & Myers, Reference Sanders and Myers2004; Sanders & Nagy, Reference Sanders and Nagy2000; Ward & Kopito, Reference Ward and Kopito1994), and eukaryotic cells have evolved elaborate systems to cope with the hazards of error-prone membrane protein synthesis and assembly. Nascent membrane proteins are subject to the scrutiny of a variety folding-sensor proteins within the ER-associated folding (ERAF) and ER-associated degradation (ERAD) pathways (comprehensively reviewed in (Vembar & Brodsky, Reference Vembar and Brodsky2008)). Collectively, these processes are referred to as ER-folding quality control (Fig. 6). As a consequence of their residence in the ER membrane, the cytosolic, lumenal, and TM portions of nascent membrane proteins are interrogated by distinct sets of quality control machinery in each of these compartments (Brodsky & Skach, Reference Brodsky and Skach2011; Buchberger et al. Reference Buchberger, Bukau and Sommer2010). Gross conformational defects in the soluble domains of membrane proteins (or perhaps the misincorporation of TM helices) may result in the inappropriate exposure of hydrophobic patches, which are likely recognized by soluble molecular chaperones such as Hsp70 in the cytosol and the Hsp70 homolog BiP in the lumen (Buchberger et al. Reference Buchberger, Bukau and Sommer2010; Meacham et al. Reference Meacham, Lu, King, Sorscher, Tousson and Cyr1999; Otero et al. Reference Otero, Lizak and Hendershot2010). ER quality control machinery also uses N-linked glycosylation markers to monitor the folding process, wherein nascent membrane proteins enter the kinetically controlled calnexin/calreticulin chaperone cycle (Ellgaard & Helenius, Reference Ellgaard and Helenius2003; Hebert & Molinari, Reference Hebert and Molinari2012; Sanders & Myers, Reference Sanders and Myers2004; Vembar & Brodsky, Reference Vembar and Brodsky2008). It is less clear how defectively folded TM domains are recognized (Houck & Cyr, Reference Houck and Cyr2012). However, a number of proteins potentially capable of recognizing defectively assembled TM domains have been identified including the E3 ubiquitin ligase HRD1 (Sato et al. Reference Sato, Schulz, Do and Hampton2009), the rhomboid homolog Derlin-1 (Sun et al. Reference Sun, Zhang, Gong, Geng, Drain and Frizzell2006), UDP-glucose: glycoprotein glucosyltransferase (Dedola et al. Reference Dedola, Izumi, Makimura, Seko, Kanamori, Sakono, Ito and Kajihara2014; Taylor et al. Reference Taylor, Ferguson, Bergeron and Thomas2004), and the translocating-chain-associated membrane protein (TRAM) (Tamborero et al. Reference Tamborero, Vilar, Martinez-Gil, Johnson and Mingarro2011). Calnexin itself has also been reported to directly sense defective TM domains in membrane proteins in a manner that is independent of its ability to recognize glycans (Cannon & Cresswell, Reference Cannon and Cresswell2001; Fontanini et al. Reference Fontanini, Chies, Snapp, Ferrarini, Fabrizi and Brancolini2005; Swanton et al. Reference Swanton, High and Woodman2003). Recognition of incompletely folded or misfolded proteins in which natively buried trafficking sequence motifs are exposed also plays a key role in ER retention or Golgi-to-ER retrieval (Geva & Schuldiner, Reference Geva and Schuldiner2014; Lee et al. Reference Lee, Miller, Goldberg, Orci and Schekman2004; Michelsen et al. Reference Michelsen, Yuan and Schwappach2005; Teasdale & Jackson, Reference Teasdale and Jackson1996; Yamamoto et al. Reference Yamamoto, Fujii, Toyofuku, Saito, Koseki, Hsu and Aoe2001). The biosynthesis of most membrane proteins is typically governed by interactions with some combination of the folding sensors described above. Protein quality control pathways are not typically ‘one set fits all’ (Buchberger et al. Reference Buchberger, Bukau and Sommer2010). Rather, different client proteins are engaged by different components and subsystems of quality control based on their physiochemical properties and the cellular physiological state (Eletto et al. Reference Eletto, Maganty, Eletto, Dersh, Makarewich, Biswas, Paton, Paton, Doroudgar, Glembotski and Argon2012; Jung et al. Reference Jung, Coe and Michalak2011; Kanehara et al. Reference Kanehara, Xie and Ng2010; Maattanen et al. Reference Maattanen, Kozlov, Gehring and Thomas2006; Molinari et al. Reference Molinari, Eriksson, Calanca, Galli, Cresswell, Michalak and Helenius2004). For instance, the conformational equilibrium of CFTR is known to be surveyed and adjusted by at least three different components of protein folding quality control: (1) its large cytosolic domain interacts with cytosolic heat-shock protein (HSP) chaperones (Hsp40, Hsp70, and Hsp90); (2) the luminal/extracellular and TM domains of the protein interact with the components of the calnexin cycle; and (3) small sequence motifs are recognized by the gatekeepers of the ER-to-Golgi transport pathway, leading to ER retention (Farinha et al. Reference Farinha, Matos and Amaral2013).
Fig. 6. ER quality control in eukaryotic cells. This diagram depicts the interplay between the ERAF and ERAD pathways within the pathway of membrane protein biosynthesis, folding, and trafficking. Nascent α-helical membrane proteins are cotranslationally inserted into the ER membrane as they come off the ribosome. The proteins then engage in quality control through numerous interactions with chaperones and folding sensors in the ERAF pathway. Nascent proteins that pass quality control are eventually exported to their destination membranes by way of the Golgi apparatus. Proteins that fail to achieve their native fold are eventually extruded from the ER membrane into the cytoplasm and polyubiquitinated. These proteins are ultimately disposed of by proteasomes or form aggregates that may be degraded or might ultimately elicit cellular toxicity.
The collective interrogation of nascent membrane proteins by folding sensors leads to one of two fates for each client protein (Brodsky, Reference Brodsky2012; Merulla et al. Reference Merulla, Fasana, Solda and Molinari2013; Olzmann et al. Reference Olzmann, Kopito and Christianson2013). Proteins deemed to be correctly and fully assembled by quality control are exported to their intended cellular compartment by way of the secretory pathway. Alternatively, proteins that fail to achieve a native fold in a timely fashion are retrotranslocated into the cytoplasm and polyubiquitinated, leading either to degradation by the proteasome or to formation of intracellular inclusions, as discussed below. The precise identity of the retrotranslocon (also referred to as the disolocon) has long been debated. It is quite possible that there is more than one protein or protein complex, including the Sec61 translocon itself, capable of serving as a retrotranslocon for various client proteins (Brodsky, Reference Brodsky2012; Merulla et al. Reference Merulla, Fasana, Solda and Molinari2013; Olzmann et al. Reference Olzmann, Kopito and Christianson2013). The genetic, cellular, and structural details of the ERAF and ERAD protein networks remain an active and fascinating area of research that can be expected to yield key insights into the molecular basis of a number of misfolding diseases. Closely aligned with ERAF and ERAD is the cellular ‘unfolded protein response’ (UPR) system that activates to relieve the burden of misfolding and/or excess nascent protein on QC pathways and the ubiquitin–proteasome systems (Bence et al. Reference Bence, Sampat and Kopito2001; Buchberger et al. Reference Buchberger, Bukau and Sommer2010; Fortun et al. Reference Fortun, Li, Go, Fenstermaker, Fletcher and Notterpek2005).
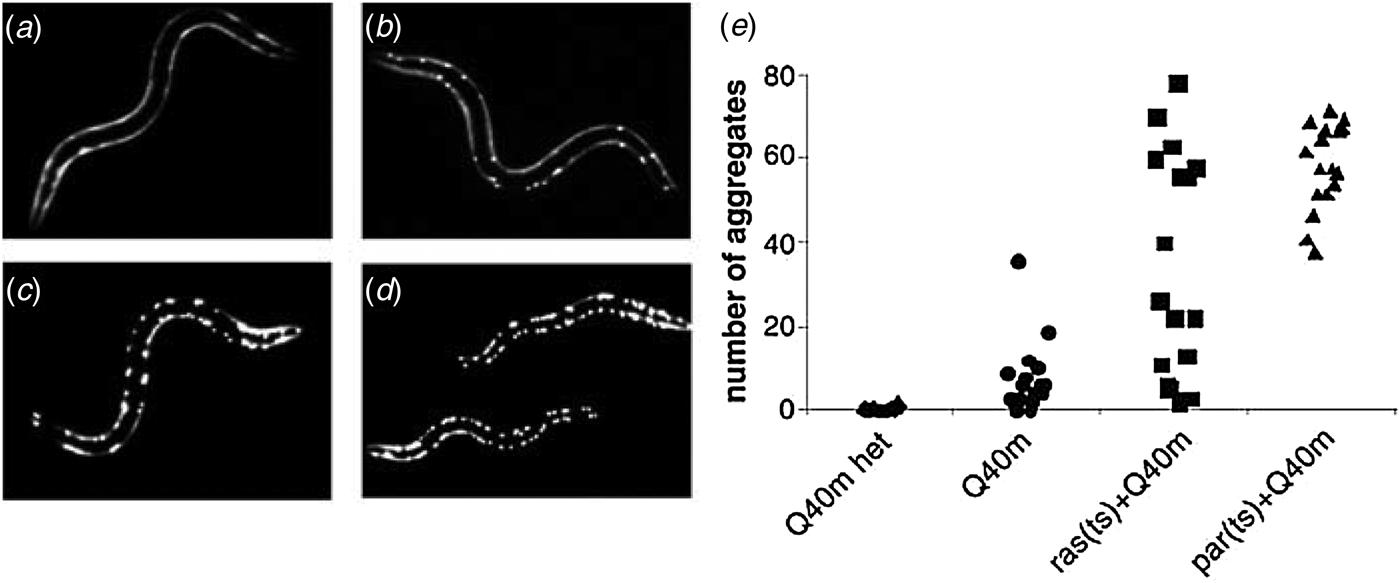
Perhaps the most obvious outcome of the misfolding of α-helical membrane proteins is the loss of native protein function associated with a reduction in the efficiency of trafficking to the intended cellular compartment (Cheng et al. Reference Cheng, Gregory, Marshall, Paul, Souza, White, O'Riordan and Smith1990; Naef & Suter, Reference Naef and Suter1999; Sanders et al. Reference Sanders, Ismail-Beigi and Mcenery2001; Sung et al. Reference Sung, Schneider, Agarwal, Papermaster and Nathans1991; Wiseman et al. Reference Wiseman, Koulov, Powers, Kelly and Balch2007a). In some cases, the pathogenesis arising as a result of misfolding seems to be due almost exclusively to the absence of functional protein in its native cellular compartment. For instance, the loss of CFTR channel function and related pathophysiology seems to be the most common outcome of the many genetic aberrations that cause CF. However, the toxic accumulation of excess misfolded membrane proteins may also represent a pathogenic stressor in some cases. Failure by the cell to fold or dispose of immature protein results in accumulation of the misfolded protein as aggregates (Kopito, Reference Kopito2000), usually following retrotranslocation of the misfolded protein into the cytosol. Although chaperones and proteasome components often colocalize with protein inclusions (Garcia-Mata et al. Reference Garcia-Mata, Bebok, Sorscher and Sztul1999; Wigley et al. Reference Wigley, Fabunmi, Lee, Marino, Muallem, Demartino and Thomas1999), the aggregates themselves tend to be relatively homogenous in composition (Rajan et al. Reference Rajan, Illing, Bence and Kopito2001). Nevertheless, intracellular aggregation of one protein can sometimes trigger the precipitation of other aggregation-prone proteins (Gidalevitz et al. Reference Gidalevitz, Ben-Zvi, Ho, Brignull and Morimoto2006) as a result of competition between multiple proteins for shared components of the ERAD and ERAF pathways (Garcia-Mata et al. Reference Garcia-Mata, Gao and Sztul2002; Gidalevitz et al. Reference Gidalevitz, Kikis and Morimoto2010). This principle has been elegantly demonstrated through a series of experiments involving genetically modified Caenorhabditis elegans, where the expression of temperature-sensitive variants of various soluble proteins was found to enhance the aggregation of fluorescent polyglutamine repeat proteins under ambient conditions (Fig. 7) (Gidalevitz et al. Reference Gidalevitz, Ben-Zvi, Ho, Brignull and Morimoto2006).
Fig. 7. Finite capacity of cellular quality control. The trans-effects of protein misfolding were examined using genetically modified Caenorhabditis elegans. Fluorescence images are shown of larvae carrying one (Q40 m het, a) or two (Q40 m, b) copies of a fluorescently-labelled, aggregation-prone polyglutamine repeat protein at permissive growth temperatures. Images are also shown of Q40 m larvae carrying temperature-sensitive mutations in the ras (c) and paramyosin (d) proteins grown at permissive temperatures. (e) Quantification of visible polyglutamine aggregates in these organisms demonstrates that the expression of other unstable proteins (i.e. mutant ras or paramyosin) can prompt the aggregation of an otherwise soluble protein (Q40 m), presumably due to competition between the two proteins for shared components of the ERAF pathway (Figure from Gidalevitz et al. Reference Gidalevitz, Ben-Zvi, Ho, Brignull and Morimoto2006).
The mechanisms by which eukaryotic cells cope with the burden of accumulated misfolded membrane proteins are currently the subject of intense investigation. Such mechanisms principally include the sequestration of misfolded and extruded membrane proteins into specialized subcellular compartments (Sontag et al. Reference Sontag, Vonk and Frydman2014). Depending on the identity and ultimate fate of the misfolded protein, it is targeted to one or more subcellular quality control compartments which include Q-bodies (Escusa-Toret et al. Reference Escusa-Toret, Vonk and Frydman2013), the juxtanuclear quality control (JUNQ) (Kaganovich et al. Reference Kaganovich, Kopito and Frydman2008), the insoluble protein deposit (IPOD; Kaganovich et al. Reference Kaganovich, Kopito and Frydman2008), aggresome-like induced structures (ALIS) (Szeto et al. Reference Szeto, Kaniuk, Canadien, Nisman, Mizushima, Yoshimori, Bazett-Jones and Brumell2006), or aggresomes (Johnston et al. Reference Johnston, Ward and Kopito1998). While both soluble proteins and α-helical membrane proteins are capable of forming these structures under certain conditions (Garcia-Mata et al. Reference Garcia-Mata, Bebok, Sorscher and Sztul1999, Reference Garcia-Mata, Gao and Sztul2002), aggresome formation has been characterized in considerable detail for a number of misfolding-prone α-helical membrane proteins, in particular for CFTR (Johnston et al. Reference Johnston, Ward and Kopito1998), PMP22 (Notterpek et al. Reference Notterpek, Ryan, Tobler and Shooter1999; Ryan et al. Reference Ryan, Shooter and Notterpek2002), and rhodopsin (Saliba et al. Reference Saliba, Munro, Luthert and Cheetham2002). Targeting of extruded and/or aggregated membrane proteins to aggresomes is achieved through an intricate network of cytosolic protein–protein interactions. First, poly-ubiquitinated aggregates are recognized by HDAC6 (Kawaguchi et al. Reference Kawaguchi, Kovacs, Mclaurin, Vance, Ito and Yao2003) and HSP70-bound proteins are detected by BAG3 (Gamerdinger et al. Reference Gamerdinger, Kaya, Wolfrum, Clement and Behl2011). These sensors then facilitate dynein-mediated retrograde transport of small diffusive aggregates to the microtubule-organizing center (MTOC) (Garcia-Mata et al. Reference Garcia-Mata, Bebok, Sorscher and Sztul1999; Johnston et al. Reference Johnston, Illing and Kopito2002; Kopito, Reference Kopito2000). A collapse of intermediate filaments around clusters of aggregates then leads to the formation of stable micron-scale aggresomes (Garcia-Mata et al. Reference Garcia-Mata, Gao and Sztul2002). These aggresomes are then gradually degraded by proteasomes and/or in lysosomes via autophagy (Fortun et al. Reference Fortun, Dunn, Joy, Li and Notterpek2003; Johnston et al. Reference Johnston, Illing and Kopito2002; Wigley et al. Reference Wigley, Fabunmi, Lee, Marino, Muallem, Demartino and Thomas1999). Importantly, it has been found that such sequestration and disposal mechanisms preserve the function of the secretory pathway (Garcia-Mata et al. Reference Garcia-Mata, Bebok, Sorscher and Sztul1999) and enhances cellular fitness (Escusa-Toret et al. Reference Escusa-Toret, Vonk and Frydman2013), suggesting that aggresomes may serve in a protective capacity (Kawaguchi et al. Reference Kawaguchi, Kovacs, Mclaurin, Vance, Ito and Yao2003; Kopito, Reference Kopito2000; Sontag et al. Reference Sontag, Vonk and Frydman2014). Nevertheless, as these mechanisms appear to be saturable (Gidalevitz et al. Reference Gidalevitz, Ben-Zvi, Ho, Brignull and Morimoto2006), an increase in the flux of nascent α-helical membrane proteins through these degradation pathways or failure to properly complete aggregate disposal may have catastrophic consequences for the cell. It should also be noted that some of the critical degradation pathways appear to decline in efficacy with aging, a factor that may contribute many aging-related disorders (Taylor & Dillin, Reference Taylor and Dillin2011).
4.2. α-Helical membrane proteins at the proteostasis boundary
Efforts to understand the linkage between protein stability and cellular proteostasis represent a frontier bridging protein biophysics with systems biology. Simplistic models are beginning to reveal the mechanisms by which the physical chemistry of protein folding gives rise to the collective functions and dysfunctions associated with cellular proteomes. Recent studies have provided reasonable predictions of the folding energetics of soluble proteins directly from the amino acid sequence (Ghosh & Dill, Reference Ghosh and Dill2009). Genomic surveys using this model predict that the cooperative unfolding of a few unstable proteins can give rise to a ‘proteostasis catastrophe’ at elevated temperatures, which coincides with the thermal cell death temperatures of bacteria, yeast, and nematodes (Ghosh & Dill, Reference Ghosh and Dill2010). Moreover, physical limitations imposed on cells by the collective instability of the proteome, the rates of biochemical reactions, and the diffusion rates of soluble proteins within the cell also seem to account for growth rates and cellular protein concentrations (Dill et al. Reference Dill, Ghosh and Schmit2011). These observations suggest a prominent role for protein stability in organismal fitness.
Bridging the knowledge gap between the energetics of protein (mis)folding and the functionality of cellular protein–protein interaction networks represents a considerable challenge. Nevertheless, a promising formalism has emerged from characterizations of transthyretin (TTR), a secreted soluble tetramer that causes systemic amyloid disease when mutated. Exhaustive folding measurements revealed an empirical relationship between the effects of pathogenic mutations on the kinetic and thermodynamic stability of TTR and both the efficiency with which the proteins are secreted by mammalian cells and the inherent amyloidogenicity of the variants (Sekijima et al. Reference Sekijima, Wiseman, Matteson, Hammarstrom, Miller, Sawkar, Balch and Kelly2005). Furthermore, it was also found that secretion efficiency is highly dependent on the functionality of cell-specific chaperone machinery as well as the concentration of native TTR ligands, which act as a chemical chaperones in the secretory pathway. Wiseman et al. later constructed a minimal model describing the linkage between folding and export (FoldEx) by simplifying the relevant interactions in the ERAD and ERAF pathways using Michealis–Menten formalism and estimated rate constants for the relevant protein–protein interactions (Wiseman et al. Reference Wiseman, Powers, Buxbaum, Kelly and Balch2007b) (Fig. 8a). Strikingly, this model was able to account for the observed relationships between the conformational stability and observed secretion efficiency of both TTR and bovine pancreatic trypsin inhibitor (BPTI) (Kowalski et al. Reference Kowalski, Parekh, Mao and Wittrup1998a, Reference Kowalski, Parekh and Wittrupb), which suggests that the principles of physical chemistry may offer utility in efforts to rationalize the elaborate cellular processes encompassed by cellular quality control. Among the concepts emerging from this model is that of a ‘proteostasis boundary’ (Powers et al. Reference Powers, Morimoto, Dillin, Kelly and Balch2009; Roth & Balch, Reference Roth and Balch2011; Fig. 8b). This concept suggests that the concentrations and activities of chemical and molecular chaperones within the cell define a ‘minimal export threshold’ that corresponds to a minimal degree of conformational stability required for proteins to pass cellular quality control. According to this model, pathological misfolding may occur when a mutation increases the misfolding rate or decreases the folding rate and/or decreases thermodynamic stability to an extent that falls below the minimal export threshold. Such variants with non-permissive conformational energetics may be targeted to ERAD, which may decrease the yield of functional protein, destabilize interaction partners, and potentially leads to the formation of cytotoxic aggregates. Together, these advances provide a general framework that may ultimately provide a means to quantitatively assess the linkage between the conformational equilibrium of a protein and its cellular fate.
Fig. 8. Physical chemistry of folding and export. (a) A schematic illustration of a simplified model for FoldEx of nascent proteins from the ER. Secreted proteins are synthesized by the translocon (T), and the nascent protein (U) either productively forms the native conformation (F) (pathway 8), or is entered into a chaperone (C) binding cycle (pathway 3). Hydrolysis of ATP is coupled to conformational changes that release the nascent protein (pathways 5 and 6) in order to enable productive folding (pathway 8) or misfolding (pathway 13). Proteins that achieve the native fold are recognized by the export machinery (E) and exported from the ER (pathways 9 and 10). However, proteins that misfold or fail to fold quickly are either reengaged by chaperones (pathways 7 and 14) or are recognized by the retrotranslocon machinery (pathways 11 and 15) and targeted for degradation. Simplification of the relevant interactions using Michaelis–Menten formalism and estimation of relevant rate constants (summarized in box) recapitulates the observed relationships between the conformational stability and export of TTR and BPTI. (Figure from Wiseman et al. Reference Wiseman, Powers, Buxbaum, Kelly and Balch2007b) (b) Simplification of the FoldEx model suggests that cell-specific expression and activities associated with ERAD and ERAF components are capable of handling proteins (different proteins represented by green) with permissible combinations of thermodynamic stability, folding rates, and misfolding rates as indicated by the cellular proteostasis boundary (purple). Proteins that fall within the proteostasis boundary are produced and degraded normally. However, destabilized variants (red) may breach this boundary (left), which can lead to the saturation of quality control and pathogenic misfolding. Because folding and export hinge on protein–protein interactions (connecting lines), destabilized variants may either directly destabilize interaction partners or indirectly destabilize competing quality control substrates (right). The destabilized variant and its interaction partners may then saturate quality control, prompt pathogenic misfolding, and induce cellular stress (Figure from Powers et al. Reference Powers, Morimoto, Dillin, Kelly and Balch2009).
The generality of these models suggests that they may eventually provide clarity to the physical limitations governing the cellular trafficking and misfolding of α-helical membrane proteins. However, rationalizing the proteostasis of membrane proteins may prove more difficult than TTR and BPTI due to the fact that significantly less is known about the conformational energetics of proteins in cellular membranes or the physical nature of their interactions with the quality control machinery. Moreover, it may be necessary to factor in the physiochemical properties of the membrane to achieve a useful proteostatic model for membrane proteins. For instance, if curvature is indeed a significant factor that influences the conformational stability of membrane proteins (Brown, Reference Brown2012), then the negative membrane curvature experienced by some proteins as they traffic through some organelles may represent a destabilizing influence on proteins that have evolved to maintain functional conformations in a destination membrane with positive curvature (Fig. 9a and b). Additionally, given that the lipid composition of cellular organelles can vary dramatically (van Meer et al. Reference Van Meer, Voelker and Feigenson2008), the conformational energetics of α-helical membrane proteins and their capacity for export may be highly dependent on the compatibility of the native conformation with the lipid compositions of the membranes that comprise each cellular compartment (Fig. 9c and d; Lundbaek et al. Reference Lundbaek, Andersen, Werge and Nielsen2003). For instance, the structural integrity of the β 2 adrenergic receptor depends on the cholesterol concentration (Zocher et al. Reference Zocher, Zhang, Rasmussen, Kobilka and Muller2012), yet the cholesterol concentration is known to be low in the ER where quality control takes place. The degree to which these factors impact the partitioning of nascent α-helical membrane proteins between the ERAF and ERAD pathways is a largely unexplored frontier. Additional biophysical studies of the effects of membranes, metabolites, and chaperones on misfolding-prone α-helical membrane proteins will be needed to formulate semi-quantitative models of the proteostasis boundary for α-helical membrane proteins.
Fig. 9. Potential influence of membrane curvature and lipid composition on the conformational equilibrium of α-helical membrane proteins. A given membrane protein may experience a variety of different membrane curvatures and lipid compositions as it traffics through the membranes of various organelles and transport vesicles. Cartoons depict the potential influences of membrane curvature and cholesterol content on protein stability. (a) For this hypothetical membrane protein the native conformation (green) is favored over unfolded conformations (yellow) in membranes with positive curvature. (Other membrane proteins may have the opposite preference.) (b) For this same hypothetical protein, negative membrane curvature destabilizes the native conformation relative to unfolded conformations. (c) For this hypothetical protein the native conformation is favored in membranes containing high concentrations of cholesterol and is less stable in membranes than contain lower cholesterol (d). (Other membrane proteins may exhibit an opposite trend.)
4.3. Pharmacological rescue of membrane proteins from misfolding and mistrafficking
Current perspectives on misfolding and proteostasis suggest several avenues for the therapeutic rescue of destabilized mutant proteins (Ong & Kelly, Reference Ong and Kelly2011; Powers et al. Reference Powers, Morimoto, Dillin, Kelly and Balch2009). The most obvious strategies involve increasing the conformational stability or suppressing misfolding and aggregation of the mutant protein. Indeed, it has long been appreciated that decreasing growth temperatures, which may increase conformational stability and suppress protein aggregation in the cell, can afford partial rescue of pathogenically misfolded membrane protein variants (Denning et al. Reference Denning, Anderson, Amara, Marshall, Smith and Welsh1992). Stabilization-based approaches have been validated by the rescue of the cellular trafficking of a number of misfolded α-helical membrane proteins in the presence of non-specific stabilizing osmolytes, including trimethylamine N-oxide (TMAO), glycerol, and dimethyl sulfoxide (DMSO; Brown et al. Reference Brown, Hong-Brown and Welch1997; Robben et al. Reference Robben, Sze, Knoers and Deen2006; Sato et al. Reference Sato, Ward, Krouse, Wine and Kopito1996; Tamarappoo & Verkman, Reference Tamarappoo and Verkman1998). However, the non-specific effects of these compounds on cellular proteostasis as well as the high concentration of osmolytes required to effectively stabilize proteins undermines their potential therapeutic utility. On the other hand, small molecules that bind with high affinity and specificity to native conformations are also capable of rescuing misfolded proteins, typically at much lower concentrations (Yu et al. Reference Yu, Sawkar and Kelly2007). Thus, for loss of function mutations that cause misfolding, low concentrations of a ligand, an agonist, or even an antagonist can facilitate a partial rescue of activity (Fan et al. Reference Fan, Ishii, Asano and Suzuki1999; Ficker et al. Reference Ficker, Obejero-Paz, Zhao and Brown2002; Morello et al. Reference Morello, Salahpour, Laperriere, Bernier, Arthus, Lonergan, Petaja-Repo, Angers, Morin, Bichet and Bouvier2000; Petaja-Repo et al. Reference Petaja-Repo, Hogue, Bhalla, Laperriere, Morello and Bouvier2002; Sawkar et al. Reference Sawkar, Cheng, Beutler, Wong, Balch and Kelly2002). Intriguingly, small molecule mediated rescue of proteins targeted for degradation suggests that conformers recognized as non-native by cellular quality control must be capable of sampling near-native, binding-competent conformations early in the secretory pathway (Fig. 10a). These near-native states likely compete with the non-native states that feed kinetically irreversible misfolding pathways (Fig. 10a–c). Thus, the influence of these compounds can be attributed to their preferential binding to and/or stabilization of the native and near-native states relative to those recognized by quality control (Bolen & Rose, Reference Bolen and Rose2008; Brown et al. Reference Brown, Hong-Brown and Welch1997; Perlmutter, Reference Perlmutter2002; Welch & Brown, Reference Welch and Brown1996), which enhances both the population and the lifetime of native conformers and facilitates their escape from the ER.
Fig. 10. Influence of pharmacological chaperones on the conformational equilibrium of multi-pass α-helical membrane proteins. (a) Scheme depicting the influence of pharmacological chaperones on the folding of nascent multi-pass α-helical membrane proteins. Nascent proteins are initially inserted into the ER membrane, where they assume a partially folded intermediate ensemble. Funneling of these intermediate ensembles through the biological folding pathway results in the formation of the correctly folded protein, which is competent for export from the ER. However, failure to do so in an efficient or timely manner may lead to a loss of the nascent protein through the ERAD pathway. The kinetics and thermodynamic of the folding transition are sensitive to the influence of pharmacological chaperones, which selectively stabilize the native conformation and/or the rate limiting transition state for folding. (b) The partitioning of the nascent protein between folding and ERAD is illustrated with an energy diagram. Unfolded conformations of nascent wild-type membrane protein (yellow) kinetically partition between the folding pathway and the ERAD pathway. Nascent proteins must overcome a rate-limiting energy barrier for folding (‡) to achieve the native conformation (green) prior to recognition by components of the ERAD pathway. (c) The incorporation of a pathogenic mutation within a TM helix (red circle) destabilizes the native conformation and/or the rate-limiting transition state for folding, which decreases the rate and efficiency of folding of the mutant protein. The effects of the mutation (red) may be partially offset in the presence of a pharmacological chaperone (blue) that preferentially binds and stabilizes the native conformation and/or the rate-limiting transition state for folding. (d) Pathogenic variants with incorrect topologies may be kinetically isolated from the native folding pathway. Reorientation of the TM helices with respect to the membrane may be outpaced by the association of the nascent protein with ERAD components. Thus, the influence of pharmacological chaperones on the conformational energy landscapes may be insufficient to rescue nascent membrane proteins with aberrant topologies.
Compounds capable of restoring the cellular trafficking and/or native function to pathogenic variants are often referred to as ‘pharmacological chaperones’. A number of pharmacological chaperones capable of preventing the misfolding of soluble proteins have progressed through clinical trials for the treatment of misfolding diseases including familial amyloid polyneuropathy (Berk et al. Reference Berk, Suhr, Obici, Sekijima, Zeldenrust, Yamashita, Heneghan, Gorevic, Litchy, Wiesman, Nordh, Corato, Lozza, Cortese, Robinson-Papp, Colton, Rybin, Bisbee, Ando, Ikeda, Seldin, Merlini, Skinner, Kelly, Dyck and Diflunisal Trial2013), Gaucher disease (Yu et al. Reference Yu, Sawkar and Kelly2007), and Fabry disease (Fan et al. Reference Fan, Ishii, Asano and Suzuki1999). With regard to diseases of membrane protein misfolding, pharmacological rescue of pathogenic CFTR variants represents a long-standing goal. Several CFTR ‘correctors,’ which mend defective CFTR trafficking, and ‘potentiators,’ which increase the open-state probability of the mature channel, have been identified and entered into clinical trials (Eckford et al. Reference Eckford, Ramjeesingh, Molinski, Pasyk, Dekkers, Li, Ahmadi, Ip, Chung, Du, Yeger, Beekman, Gonska and Bear2014; Odolczyk et al. Reference Odolczyk, Fritsch, Norez, Servel, Da Cunha, Bitam, Kupniewska, Wiszniewski, Colas, Tarnowski, Tondelier, Roldan, Saussereau, Melin-Heschel, Wieczorek, Lukacs, Dadlez, Faure, Herrmann, Ollero, Becq, Zielenkiewicz and Edelman2013; Rowe & Verkman, Reference Rowe and Verkman2013; Sampson et al. Reference Sampson, Robert, Liao, Matthes, Carlile, Hanrahan and Thomas2011; Van Goor et al. Reference Van Goor, Hadida, Grootenhuis, Burton, Cao, Neuberger, Turnbull, Singh, Joubran, Hazlewood, Zhou, Mccartney, Arumugam, Decker, Yang, Young, Olson, Wine, Frizzell, Ashlock and Negulescu2009, Reference Van Goor, Hadida, Grootenhuis, Burton, Stack, Straley, Decker, Miller, Mccartney, Olson, Wine, Frizzell, Ashlock and Negulescu2011). However, identification of a compound capable of mediating sufficient and robust rescue of the common ΔF508 CF variant remains an ongoing challenge (Rowe & Verkman, Reference Rowe and Verkman2013). Efforts to develop novel pharmacological chaperones have also targeted a number of other pathogenic membrane protein variants believed to promote misfolding including the E90K variant of gonadotropin-releasing hormone receptor (GhRHR), the cellular mistrafficking and the associated disease phenotype of which have recently been corrected in mice using both a native ligand and an agonist (Janovick et al. Reference Janovick, Stewart, Jacob, Martin, Deng, Stewart, Wang, Cornea, Chavali, Lopez, Mitalipov, Kang, Lee, Manna, Stocco, Behringer and Conn2013). Numerous other misfolding-prone membrane proteins are known to be competent for rescue by pharmacological chaperones include some 18 different GPCRs such as rhodopsin (retinitis pigmentosa), the V2R (diabetes insipidus) (Tao & Conn, Reference Tao and Conn2014), nicotinic acetylcholine receptors (Srinivasan et al. Reference Srinivasan, Henderson, Lester and Richards2014), KATP channels (Martin et al. Reference Martin, Chen, Devaraneni and Shyng2013), and the HERG potassium channel (Gong et al. Reference Gong, Jones and Zhou2006). These advances highlight the growing interest in pharmacological chaperones as a therapeutic strategy for diseases of protein folding.
Proteostasis regulators (PR), which alter the general capacity of cellular ERAD and ERAF pathways, represent an emerging alternative in cases where the development of protein-specific pharmacological chaperones may be impractical or ineffective (Balch et al. Reference Balch, Morimoto, Dillin and Kelly2008; Mu et al. Reference Mu, Ong, Wang, Balch, Yates, Segatori and Kelly2008; Powers et al. Reference Powers, Morimoto, Dillin, Kelly and Balch2009). This approach has recently shown great promise. Screening efforts have yielded a number of compounds that may offer general therapeutic utility for misfolding diseases (Calamini et al. Reference Calamini, Silva, Madoux, Hutt, Khanna, Chalfant, Saldanha, Hodder, Tait, Garza, Balch and Morimoto2012; Carlile et al. Reference Carlile, Keyzers, Teske, Robert, Williams, Linington, Gray, Centko, Yan, Anjos, Sampson, Zhang, Liao, Hanrahan, Andersen and Thomas2012; Mu et al. Reference Mu, Ong, Wang, Balch, Yates, Segatori and Kelly2008; Tardiff et al. Reference Tardiff, Jui, Khurana, Tambe, Thompson, Chung, Kamadurai, Kim, Lancaster, Caldwell, Caldwell, Rochet, Buchwald and Lindquist2013). Interestingly, the rescue afforded by PRs seems to arise through diverse biochemical mechanisms including the modulation of ER stress pathways (Ozcan et al. Reference Ozcan, Yilmaz, Ozcan, Furuhashi, Vaillancourt, Smith, Gorgun and Hotamisligil2006; Ryno et al. Reference Ryno, Wiseman and Kelly2013; Wiseman & Balch, Reference Wiseman and Balch2005), the tuning of HSP expression and activity (Calamini et al. Reference Calamini, Silva, Madoux, Hutt, Khanna, Chalfant, Saldanha, Hodder, Tait, Garza, Balch and Morimoto2012), inhibition of the proteasome (Mu et al. Reference Mu, Ong, Wang, Balch, Yates, Segatori and Kelly2008), and inhibition of ubiquitin ligases (Tardiff et al. Reference Tardiff, Jui, Khurana, Tambe, Thompson, Chung, Kamadurai, Kim, Lancaster, Caldwell, Caldwell, Rochet, Buchwald and Lindquist2013). Therefore, these compounds may represent a toolbox that can be used to address the potentially distinct cellular stressors that arise as a result of various misfolding mechanisms. However, due to the lack of specificity of such compounds, it is currently unclear what kinds of off-target effects they may elicit in the context of the human body. Nevertheless, considering the systemic nature of certain misfolding disorders, it may very well be that the therapeutic merits of these compounds outweigh potential side effects.
It has been observed that certain pathogenically misfolded variants of α-helical membrane proteins appear to be incompetent for ligand binding (Ficker et al. Reference Ficker, Obejero-Paz, Zhao and Brown2002; Kaushal & Khorana, Reference Kaushal and Khorana1994; Sung et al. Reference Sung, Schneider, Agarwal, Papermaster and Nathans1991), which may indicate that the misfolded ensembles achieved by these variants are kinetically isolated from binding-competent, near-native states. In the context of α-helical membrane proteins, one possible cause of this class of misfolding are pathogenic variants that prompt cotranslational misfolding and reside in binding-incompetent topologies, which may not readily interconvert with native topologies (Fig. 10d). In this case, pharmacological chaperones are unlikely to be of use and modulation of the proteostasis boundary may instead represent a more effective strategy to manage the consequences of misfolding. The effects of pathogenic mutations on the biochemical function of the rescued protein must also be factored into rational therapeutic design. A pointed example was provided by a recent study demonstrating that a mutation in a sodium channel that prompts ER-retention also causes aberrant constitutive activation upon correction of folding defects (Cestèle et al. Reference Cestèle, Schiavon, Rusconi, Franceschetti and Mantegazza2013). One can imagine that, in such a case, a channel blocker-mediated pharmacological rescue might be more desirable than rescue mediated by an activating ligand. Such considerations provide the impetus for experimentally examining effects of a given pathogenic mutation on the biogenesis, folding, and function of a target α-helical membrane protein, suggesting important roles for biochemistry and structural biology in personalized medicine.
5. Conclusions and outlook
Fundamental questions involving the principles of membrane protein folding and misfolding have long remained elusive. Nevertheless, significant advances in recent years are beginning to shed light on the relationships between the biophysics of α-helical membrane protein misfolding, the role of the translocon in their biosynthesis, the nature of their interactions with components of ER quality control, the biochemical nature of the resulting cellular pathology, and the molecular basis of misfolding disease. Delineation of the effects of pathogenic mutations on membrane proteins may offer a new degree of clarity for therapeutic design. Moreover, future efforts to unite structural biology with the tools and perspectives of cellular biology, biochemistry, and chemical biology offer the potential to comprehensively elucidate the effects of pathogenic mutations at the level of systems biology. These advances and perspectives offer hope for the development of novel therapeutics that may be of use in a multitude of diseases, potentially in a genotype-specific (personalized) manner. These diseases will include not only inherited (Mendelian) disorders with simple mutation–disease relationships, but also ‘complex’ disorders such as sporadic Alzheimer's disease, various cardiovascular problems, and type 2 diabetes, where risk factors that promote membrane protein misfolding may be common contributors to disease etiology and pathology.
6. Acknowledgements
This work was supported by NIH grants RO1 DC007416, RO1 DK083187, RO1 GM106672, RO1 HL122010 and U54 GM094608. We thank Kathleen Mittendorf for helpful discussion. We apologize to the authors of related work that was not cited herein because of author oversight or because of space limitations.