Introduction
The correct identification of species or intraspecific entities must be the primary goal for the accomplishment of biodiversity surveys, biological control and pest management strategies (Rosen, Reference Rosen1986; Paterson, Reference Paterson and Zalucki1991; Mills & Kean, Reference Mills and Kean2010). Correct taxonomy is also essential to prevent, detect and respond to the invasion of a species in a new area (Douglas et al., Reference Douglas, Dang, Gill, Huber, Mason, Parker and Sinclair2009). Hence, species misidentification can hinder attempts toward their conservation or control. The inaccurate recognition of species complexes can lead to erroneous employment of pest control approaches as different species can cause variable degrees of damage (Rugman-Jones et al., Reference Rugman-Jones, Hoddle and Stouthamer2010), and can present divergent answers to control measures (Ríos-Díez & Saldamando-Benjumea, Reference Ríos-Díez and Saldamando-Benjumea2011) and patterns of resistance to pesticides (Bickford et al., Reference Bickford, Lohman, Sodhi, Ng, Meier, Winker, Ingram and Das2007).
Telchin licus (Drury), the giant sugarcane borer, is the main pest attacking sugarcane (Saccharum officinarum L.) in northeast Brazil. Primarily restricted to the north and northeast regions, the giant sugarcane borer was recently reported in southeast Brazil, in the state of São Paulo (Almeida et al., Reference Almeida, Dias Filho and Arrigoni2007), the main sugarcane producer area in Brazil, raising concerns about its control. It has been suggested that T. licus was introduced into sugarcane-producing areas through the transport of ornamental plants (Almeida et al., Reference Almeida, Dias Filho and Arrigoni2007), as already reported for the castniid Paysandisia archon (Burmeister) in Europe (Sarto i Monteys & Aguilar, Reference Sarto i Monteys and Aguilar2005). However, the origin of the population of T. licus currently found in São Paulo is still an issue under investigation.
Telchin licus is part of a species complex that includes Telchin syphax (Fabricius) and Telchin atymnius (Dalman), new combinations after Erythrocastnia and Castniomera were synonymized with Telchin (Gonzalez & Cock, Reference Gonzalez and Cock2004; Moraes & Duarte, Reference Moraes and Duarte2009). There is scarce morphological variation among species in the T. licus complex, and taxa are commonly identified based on their wing colour pattern, despite the fact that several Neotropical Castniinae are mimetics (Miller, Reference Miller1986). Telchin licus and T. atymnius share some sympatry in their distribution range, and both are known to feed on S. officinarum and Musaceae (Moraes & Duarte, Reference Moraes and Duarte2009). Yet, their accurate identification becomes even harder as 12 geographically differentiated subspecies are recognized for T. licus, four of them in Brazil: Telchin licus licus (Drury), Telchin licus albomaculata (Houlbert), Telchin licus laura (Druce) and Telchin licus rubromaculata (Houlbert) (Lamas, Reference Lamas1995). These subspecies were proposed mainly based on their geographical distribution and subtle differences in wing colour. Little is known either on their limits or on the phylogenetic relationships among them.
Therefore, considering the sparse knowledge of the group and the economic importance of T. licus, there is an urgent need for a reliable source of information that would allow the proper identification of species/subspecies belonging to the T. licus species complex. Molecular markers, such as mitochondrial DNA (mtDNA), are excellent candidates to solve the taxonomic issues faced in the identification of species of the T. licus complex, as they have been successfully used to solve taxonomic problems where morphological data is uninformative, aiding in the definition of species boundaries and in the identification of the origin of new populations (Sperling & Hickey, Reference Sperling and Hickey1994; Peña et al., Reference Peña, Wahlberg, Weingartner, Kodandaramaiah, Nylin, Freitas and Brower2006; Behere et al., Reference Behere, Tay, Russell, Heckel, Appleton, Kranthi and Batterham2007; Porreta et al., Reference Porreta, Canestrelli, Bellini, Celli and Urbanelli2007; Norgate et al., Reference Norgate, Chamings, Pavlova, Bull, Murray and Sunnucks2009). Indeed, a region of the animal mtDNA was proposed as a ‘DNA barcode’ for species diagnoses and delimitation (Hebert et al., Reference Hebert, Cywinska, Ball and DeWaard2003). The benefit of using mtDNA lays on its intrinsic characteristics, such as the high rate of evolutionary change, mostly maternal inheritance, high genetic polymorphism among conspecifics and the availability of universal primers (Avise, Reference Avise1986; Moritz et al., Reference Moritz, Dowling and Brown1987; Simon et al., Reference Simon, Frati, Beckenbach, Crespi, Liu and Flook1994).
The objectives of this study were to use mtDNA to (i) investigate the origin of specimens of T. licus sampled from the state of São Paulo by using population genetics methods in order to understand the dissemination of this important pest throughout Brazil, and (ii) assess the effectiveness of mtDNA, and specifically of the ‘barcode’ region, in diagnosing T. licus taxonomic units and in recovering the relationships among them.
Material and methods
Samples
Specimens of T. licus were sampled from five sites in northeast, southeast and central Brazil in 2009 and 2010. Larvae were collected from sugarcane plants from Coruripe, AL (CO_AL, n = 10), São Miguel, AL (SM_AL, n = 10), Limeira, SP (LI_SP, n = 10), Tangará da Serra, MT (TS_MT, n = 11) and Araporã, MG (AR_MG, n = 3) (fig. 1) and were immediately fixed in 96% ethanol. Vouchers of adults from each sampling locality were obtained by rearing larvae until pupation and later adult emergence, and were maintained in the Centro de Tecnologia Canavieira (CTC) by L. C. Almeida. Additionally, two adults of T. licus were sampled from the Amazon Forest region in the margins of Madeira River, ca. of 150 km from Porto Velho, RO (PV_RO); vouchers were preserved in the Museu de Zoologia of Universidade de São Paulo (MZUSP) (Voucher codes: C1_Mol07 and C2_Mol08). Sample sets from each locality were treated as separated populations.

Fig. 1. Map of Brazil with Telchin licus populations' origin indicated.
DNA extraction, amplification and sequencing
Genomic DNA was obtained according to the Invisorb Spin Tissue kit (STRATEC Molecular, Berlin, Germany) protocol from the last abdominal segment of each larva and from one leg of each adult. DNA extractions were stored in TE buffer at −20 °C. The mitochondrial genes cytochrome c oxidase I (cox1, ca. 1480 bp) and the subunit 6 of the nicotinamide adenine dinucleotide dehydrogenase (nad6, ca. 500 bp) were amplified using the follow primers combinations: LCO (5′ GGTCAACAAATCATAAAGATATTGG) + HCO (5′ TAAACTTCAGGGTGACCAAAAAATCA) (Folmer et al., Reference Folmer, Black, Hoeh, Lutz and Vrijenhoek1994) and Jerry (5′ CAACATTTATTTTGATTTTTTGG 3′) + PatII (5′ TCCATTACATATAATCTGCCATATTAG 3′) (Caterino et al., Reference Caterino, Reed, Kuo and Sperling2001) for cox1, and tPro-J10090 (5′ ATCWATAATCTCCAAAATTAT 3′) + ND6-N10624 (5′ GGNCCATAAAAAATATTWGT 3′) (Silva-Brandão et al., Reference Silva-Brandão, Lyra, Santos, Seraphim, Albernaz, Pavinato, Martinelli, Cônsoli and Omoto2011) for nad6. Reactions were done in a 25 μl final volume using 1 μl of total DNA, 2.0 mM of MgCl2, 40 μM of dNTPs, 0.2 μM of each primer, 1 U of GoTaq DNA Polymerase (Promega, Madison, WI, USA), and 10% of 10 × Taq buffer. The PCR program to amplify cox1 included an initial denaturation step at 95 °C for 3 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 45–47 °C for 30 s, and polymerization at 72 °C for 1.5 min, followed by an extension step at 72 °C for 10 min (Silva-Brandão et al., Reference Silva-Brandão, Freitas, Brower and Solferini2005). The amplification protocol for nad6 was as follows: an initial denaturation step at 94 °C for 5 min, 35 cycles of denaturation at 94 °C for 45 s, annealing at 45 °C for 45 s, and elongation at 60 °C for 1.5 min, followed by an extension step at 60 °C for 5 min (Silva-Brandão et al., Reference Silva-Brandão, Lyra, Santos, Seraphim, Albernaz, Pavinato, Martinelli, Cônsoli and Omoto2011). Amplicons were purified of primers and deoxynucleotides with ExoSAP-IT (GE Healthcare, Bucks, UK), and then sequenced by ABI Prism BigDye Kit protocol. Mitochondrial regions were sequenced in an ABI 3700 automated sequencer, with primers used for amplification. Sequences were analyzed with the program FinchTV v. 1.4.0 (Geospiza Inc., Seattle, WA, USA), and aligned manually using BioEdit v. 7.0.5.3 (Hall, Reference Hall1999).
Genetic distances and clustering analyses
All analyzes were applied to concatenated data of the mitochondrial genes cox1 and nad6, and genetic variation and DNA polymorphism were also estimated for each mitochondrial region separately. Standard parameters of DNA polymorphism were estimated in DnaSP v. 5.10 (Librado & Rozas, Reference Librado and Rozas2009) and in MEGA v. 5.0 (Tamura et al., Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011). The DNA polymorphism using the sliding window method was estimated for the determination of the number of segregating sites (S) across each portion of the mtDNA sequences, set with a window length = 100 and a step size = 25 (Librado & Rozas, Reference Librado and Rozas2009).
A minimum spanning network representing the genealogical relationships of haplotypes was constructed using the method of Statistical Parsimony (Templeton et al., Reference Templeton, Crandall and Sing1992) as implemented in TCS v. 2.1 (Clement et al., Reference Clement, Posada and Crandall2000), using 95% confidence.
Sequences divergence among individuals of T. licus were quantified by using the p-distance model of nucleotide substitution (Nei & Kumar, Reference Nei and Kumar2000), implemented in the program MEGA v. 5.0 (Tamura et al., Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011). The neighbor-joining (NJ) clustering algorithm (Saitou & Nei, Reference Saitou and Nei1987) was applied to graphically obtain the phenetic distance among individuals of T. licus estimated by using the p-distance model. Robustness of each branch was determined by using 1000 replicates of the non-parametric bootstrapping procedure (Felsenstein, Reference Felsenstein1985).
Concatenated data of cox1 and nad6 was used to estimate a Bayesian inference (BI) tree. The dataset was partitioned in two regions, and the program MEGA v. 5.0 (Tamura et al., Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011) was used to determine the available substitution model with the best fit to each mitochondrial region. The best fit models suggested for cox1 and nad6 sequences were HKY + G and HKY (Hasegawa et al., Reference Hasegawa, Kishino and Yano1985), respectively. The tree was estimated by using the program Beast v. 1.6.1 (Drummond & Rambaut, Reference Drummond and Rambaut2007), with two Markov Chain Monte Carlo runs for 40,000,000 generations, each MCMC starting from a random tree, using a relaxed clock and Yule speciation model, and sampling trees every 5000 generations. The posterior distribution of trees was summarized with TreeAnnotator v. 1.4.7 (Drummond & Rambaut, Reference Drummond and Rambaut2007), using a 20% burn-in and computing the maximum clade credibility tree with average branch lengths.
The effectiveness of the 5′ extremity of cox1, the proposed ‘barcode’ region (Hebert et al., Reference Hebert, Cywinska, Ball and DeWaard2003), for diagnosing taxonomic units of T. licus, was accessed by constructing a NJ tree with sequences of this fragment (ca. 640 bp), by using the nucleotide substitution model Kimura-2-Parameters (K2P) (Kimura, Reference Kimura1980) and 1000 replicates of bootstrap (Felsenstein, Reference Felsenstein1985). A minimum spanning network was also constructed using this mitochondrial region in TCS v. 2.1 (Clement et al., Reference Clement, Posada and Crandall2000) with 95% confidence.
Population structure
Within and among population divergences were compared by using standard AMOVA (Excoffier et al., Reference Excoffier, Smouse and Quattro1992), as implemented in the program Arlequin v. 3.5 (Excoffier & Lischer, Reference Excoffier and Lischer2010). Sequences of adults from Porto Velho (RO) were not analyzed due to the low number of sampled specimens. Two analyses were conducted to quantify the distribution of the molecular variation attributed to the presence of genetic structure: (i) among individuals from all populations (non-hierarchic) and (ii) among clusters recovered with NJ analysis of concatenated datasets. The degree of structure was interpreted by the Φ statistics associated with the different hierarchical levels in which the variation is distributed (Excoffier et al., Reference Excoffier, Smouse and Quattro1992). The parameters applied were: 10,000 permutations to determine significance, computed distance matrix using pairwise difference, and gamma a value equal 0. Pairwise linearized FST (Slatkin, Reference Slatkin1995) was estimated by using Arlequin v. 3.5. The hypothesis of genetic isolation by geographical distance was assessed with the Mantel test (Mantel, Reference Mantel1967), with 10,000 permutations, using Slatkin's linearized FST (Slatkin, Reference Slatkin1995) and linear geographic distances.
Results
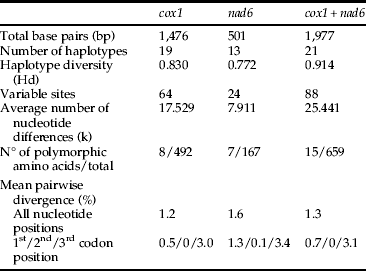
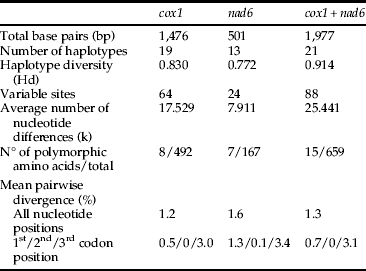
The analyzed cox1 and nad6 fragments were 1476 bp and 501 bp long, respectively (GenBank accession numbers: cox1, JQ685038–JQ685083 and nad6, JQ685084–JQ685129). The mean genetic distance among cox1 sequences was 0.012 (0–0.026). For nad6, this value was a little higher, 0.016 (0–0.036). Overall genetic distance was 0.013 (0–0.028) for concatenated datasets (table 1).
Table 1. Statistical summary of nucleotide sequences of T. licus.
The DNA polymorphism of combined datasets using the sliding window method showed sequences of the 3′ extremity of cox1 (ca. 840 bp) as the most variable, followed by sequences of nad6 and the 5′ end of cox1 (fig. 2).

Fig. 2. Number of segregating sites (S) across mitochondrial regions.
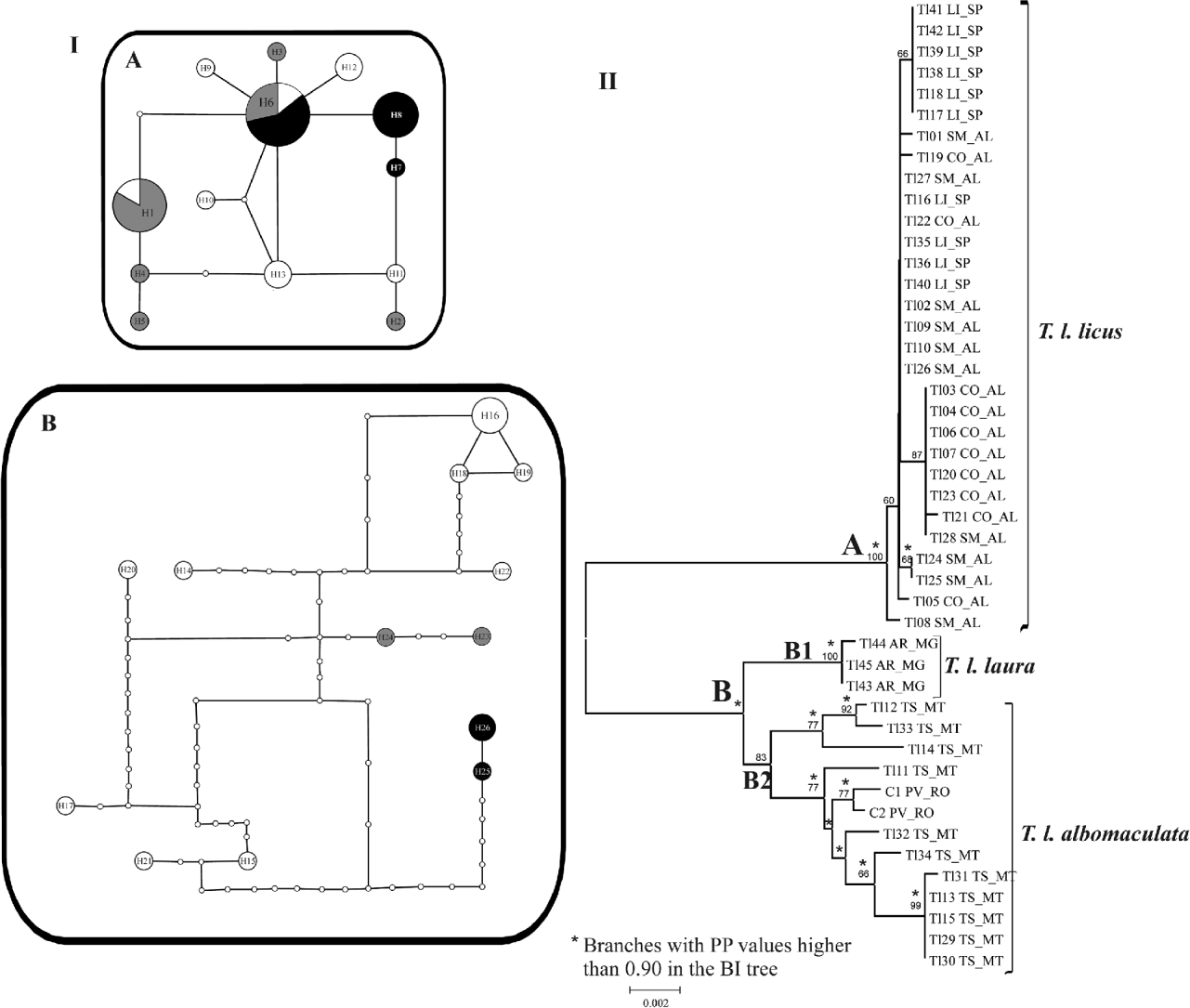
The 46 combined mtDNA sequences recovered two disconnected networks with 13 haplotypes each. Fixing the connection limit at 45 steps resulted in a joined network (data not shown). The first network (fig. 3 IA) is composed of the three sample sets from Limeira (LI_SP), Coruripe (CO_AL) and São Miguel (SM_AL). This network shows three haplotypes for samples from LI_SP (6, 7, 8), two of them exclusive occurrences for this locality, six for samples from CO_AL (1, 2, 3, 4, 5, 6) and seven for samples from SM_AL (1, 6, 9, 10, 11, 12, 13). Only haplotypes 1 and 6 are shared among sampled localities. The second network is formed by samples from Araporã (AR_MG), Tangará da Serra (TS_MT) and Porto Velho (PV_RO) (fig. 3 IB). This network shows nine exclusive haplotypes for samples from TS_MT (from 14 to 22), and two for samples from PV_RO (23, 24) and AR_MG (25, 26).
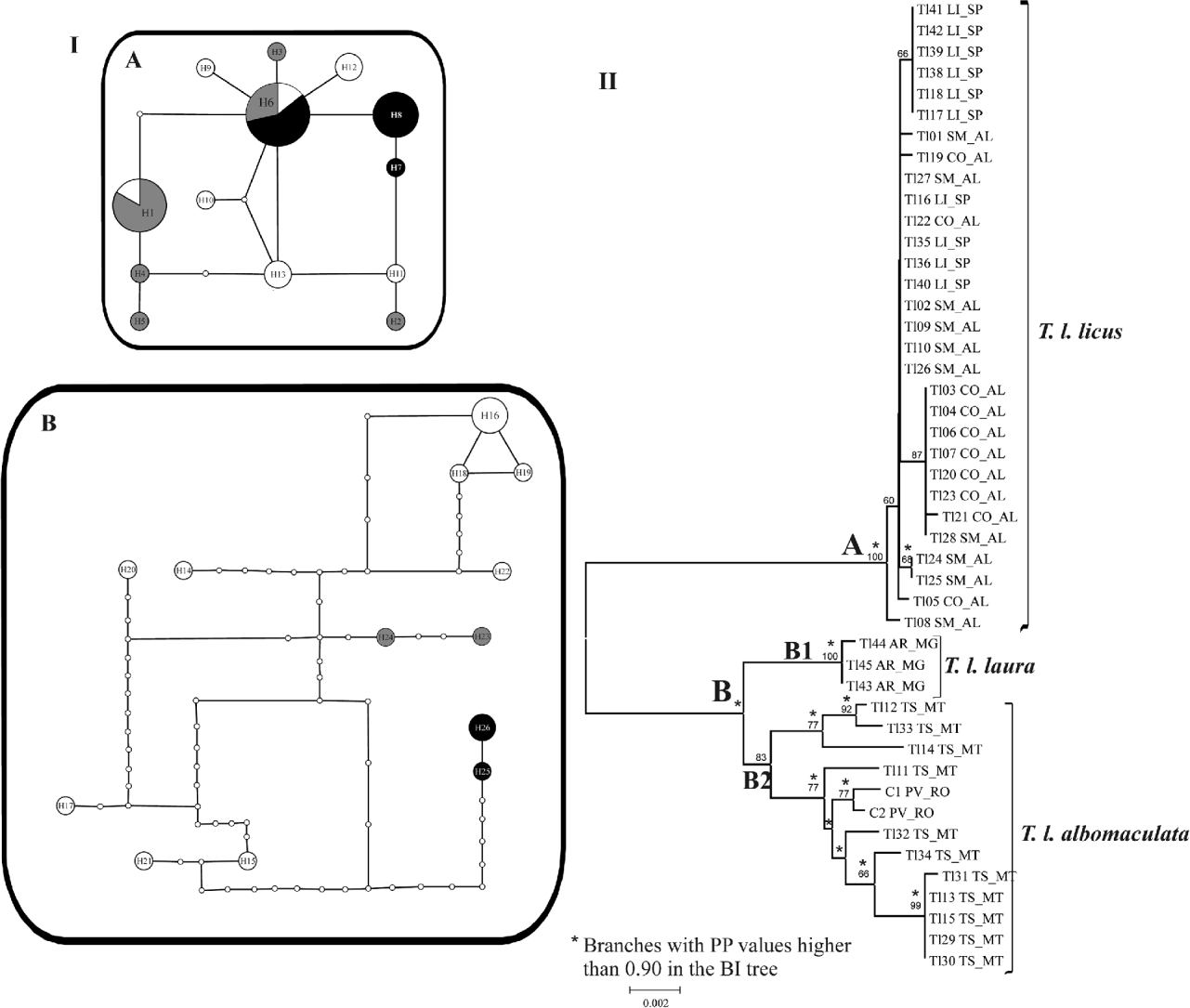
Fig. 3. (I) Minimum spanning networks for T. licus populations based on combined datasets of cox1 and nad6. A and B networks are composed of samples from clades A and B in the neighbor-joining tree. Circle areas are directly proportional to the number of individuals showing such haplotype. Small white circles indicate unsampled haplotypes. Each branch is equivalent to one base pair change. (II) Neighbor-joining tree obtained with p-distance model of nucleotide substitution of concatenated data of cox1 and nad6 sequences. Numbers on the branches indicate bootstrap values of 1000 replicates (when exceed 50%). Branches recovered by Bayesian inference (BI) with posterior probability (PP) values higher than 0.90 are indicated by an *. Sampled sites: CO_AL, Coruripe, AL; LI_SP, Limeira, SP; SM_AL, São Miguel, AL; TS_MT, Tangará da Serra, MT; AR_MG, Araporã, MG; PV_RO, Porto Velho, RO. (I (A) •, LI_SP = Limeira, SP; ![]() , CO_AL = Coruripe, AL; ○, SM_AL = São Miguel, AL; I (B) •, AR_MG = Araporã, MG;
, CO_AL = Coruripe, AL; ○, SM_AL = São Miguel, AL; I (B) •, AR_MG = Araporã, MG; ![]() , PV_RO = Porto Velho, RO; ○, TS_MT = Tangará da Serra, MT)
, PV_RO = Porto Velho, RO; ○, TS_MT = Tangará da Serra, MT)
The NJ and the BI trees clustered the samples in two main clades (fig. 3 II). The clade A is supported by high bootstrap and posterior probability (PP) values, and is formed by samples from SM_AL, LI_SP and CO_AL, with no geographical distinction. Clade B comprises two main clusters: the highly supported clade B1 is formed by samples from AR_MG, and the moderately supported clade B2 is composed by specimens from TS_MT, sampled from sugarcane fields, and PV_RO, sampled from a region of Amazon Forest. Genetic p-distance within clade A was 0.1%, 0 within B1 and 0.6% within B2. The genetic p-distance was 2.3% between clades A and B1, 2.6% between A and B2 and 1.1% between B1 and B2.
Using only sequences of the 5′ extremity of cox1 resulted in a weakly supported clade B and a paraphyletic clade B2. Hence, B2 was considered to be two subclades named C1 (with moderate bootstrap support) and C2 (showing weak bootstrap support) (fig. 4 I). There were two segregating sites restricted to samples from TS_MT, one exclusively found in samples from AR_MG, and three shared between the two localities (TS_MT and AR_MG). Sequences of this mitochondrial region alone were not sufficient to recover the clades recuperated by using the concatenated datasets of cox1 and nad6.

Fig. 4. (I) Neighbor-joining tree obtained with Kimura-2-Parameters model of nucleotide substitution of sequences of the 5′ extremity of cox1. Numbers on the branches indicate bootstrap values of 1000 replicates (when exceed 50%). (II) Minimum spanning network for T. licus populations based on sequences of the 5′ extremity of cox1. (II •, ARMG = Araporã, MG; ![]() , PVRO = Porto Velho, RO; ○, TSMT = Tangará da Serra, MT)
, PVRO = Porto Velho, RO; ○, TSMT = Tangará da Serra, MT)
Two disconnected networks were recovered with the 5′ end of cox1. The first network, composed by samples from LI_SP, CO_AL and SM_AL, only had two haplotypes (data not shown). The other network, composed by samples from AR_MG, TS_MT and PV_RO, had eight haplotypes distributed in a structure similar to the clades found with NJ analysis (fig. 4).
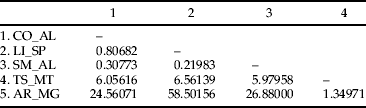
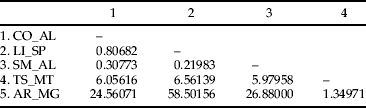
The estimated value of ΦST for T. licus populations was 0.86 (P < 0.001), and most of the variation was found among populations (table 2). The hierarchical AMOVA among clusters recovered in the NJ tree of combined datasets showed that 89.95% of variation is among groups (table 2). The higher pairwise Slatkin's value was found between populations from AR_MG and LI_SP, while the lowest genetic variation was found between populations from SM_AL and LI_SP (table 3). The correlation analysis revealed the lack of a significant association between genetic and geographic distances (Mantel test, P = 0.79).
Table 2. Analysis of molecular variance (AMOVA) (Excoffier et al., Reference Excoffier, Smouse and Quattro1992) for T. licus populations based on combined datasets of cox1 and nad6.

Table 3. Slatkin's linearized FST values (Slatkin, Reference Slatkin1995) among populations of T. licus. Sampled localities: CO_AL, Coruripe, AL; LI_SP, Limeira, SP; SM_AL, São Miguel, AL; TS_MT, Tangará da Serra, MT; AR_MG, Araporã, MG. All values are significant at significance level 0.05.
Discussion
Inaccurate identification, and definition of species boundaries for cryptic pest species or intraspecific taxa have important implications for handling biological invasions (Douglas et al., Reference Douglas, Dang, Gill, Huber, Mason, Parker and Sinclair2009; Guillemaud et al., Reference Guillemaud, Ciosi, Lombaert and Estoup2011) and can result in inadequate application of control measures (Rosen, Reference Rosen1986; Bickford et al., Reference Bickford, Lohman, Sodhi, Ng, Meier, Winker, Ingram and Das2007). The taxonomic limits of the Neotropical castniids are frequently based on wing colour pattern and geographical distribution (Moraes & Duarte, Reference Moraes and Duarte2009; Moraes et al., Reference Moraes, Duarte and Gonzalez2010), even though these traits are not always sufficient to provide a clear picture of the boundaries of these entities, or yet to produce a coherent classification in some cases.
Our analyses of mitochondrial markers indicated a high genetic variability among populations of T. licus from Brazil. Indeed, overall ΦST values, paired F ST and the genealogical network analyzes pointed out to a high genetic structure among populations. This structuring is in agreement with the occurrence of at least four subspecies of T. licus in Brazil (Lamas, Reference Lamas1995). Considering Lamas’ (Reference Lamas1995) checklist and pictures of type specimens, samples from LI_SP, CO_AL and SM_AL (clade A in fig. 3) were identified as Telchin licus licus (picture presented in Houlbert (Reference Houlbert1918) as Castnia licoides, Pl. CDXLIV), although its type specimen had been originally sampled from the state of Santa Catarina (Lamas, Reference Lamas1995). We considered the sample set from TS_MT and PV_RO (clade B2 in fig. 3) as Telchin licus albomaculata (picture presented in Houlbert (Reference Houlbert1918) as Castnia albomaculata, Pl. CDXLIV). The type specimen of T. licus albomaculata is from Amazônia, Brazil, nearby to Peru (Lamas, Reference Lamas1995). Lamas' (Reference Lamas1995) checklist presents the name Telchin licus laura for Mato Grosso (MT), but this name is currently used for samples from Minas Gerais (MG), and samples from AR_MG were named as such (clade B1 in fig. 3) (photo of the type specimen is available online at http://www2.nrm.se/en/lep_nrm/l/castnia_laura.html). There are indeed two possible hypotheses concerning the identification of this subspecies: (i) the geographical range of T. licus laura can be broader than initially inferred from the original type description and extends the limits beyond Mato Grosso, reaching the state of Minas Gerais, or (ii) populations of T. licus laura from Mato Grosso and Minas Gerais are indeed distinct entities. In this case, the name could not be applied, and a new name should be proposed for the population of Minas Gerais. This second possibility should only be considered after evaluation of samples of T. licus laura from Mato Grosso. Nevertheless, the first hypothesis is more likely as there are three specimens identified as T. licus laura in the collection of the Department of Zoology of Universidade Federal do Paraná (OM): one specimen 20-ii-1967 from Uberlândia (MG), one 02-i-1984 from Chapada dos Guimarães (MG) and one 24-i-1968 from Brasília (DF). Additionally, a specimen of T. licus laura was recently reported in Paraguay (Rios & Gonzalez, Reference Rios and Gonzalez2011).
Adults of T. licus laura are easily differentiated from adults of T. licus licus and T. licus albomaculata by the presence of an additional submarginal orange patch in the tornus of dorsal and ventral forewings. On the other hand, adults of T. licus licus and T. licus albomaculata are morphologically indistinguishable; however, our data indicate they are genetically isolated entities. These two subspecies belong to a special category of cryptic taxa; and, in these cases, even the most careful morphological examination does not allow for their differentiation, although they can be recognized as ‘genetical species’ (Paterson, Reference Paterson and Zalucki1991). Our data suggests a close phylogenetic relationship between T. licus laura and T. licus albomaculata, but only the inclusion of the unsampled subspecies T. licus rubromaculata will permit a broader understanding of the relationships among these subspecies.
Barcode diagnosing T. licus entities
The proposal of applying a fragment of mtDNA as animal ‘barcode’ presents two different approaches: the identification or diagnosis of species, and the description or delimitation of new species (Hebert et al., Reference Hebert, Cywinska, Ball and DeWaard2003). Most of the current criticism is driven to the second approach (DeSalle et al., Reference DeSalle, Egan and Siddall2005). Despite pitfalls of the ‘DNA barcode’ proposal, the 5′ end of cox1 has been successfully used for species diagnose in well-studied groups of Lepidoptera (Hebert et al., Reference Hebert, Penton, Burns, Janzen and Hallwachs2004; Silva-Brandão et al., Reference Silva-Brandão, Lyra and Freitas2009).
Therefore, the nomenclature proposed above to identify the genetic groups recovered in the populations of T. licus must consider the sample set from TS_MT that was recovered as paraphyletic when only the 5′ extremity of cox1 was used as input. Several reasons may result in lack of monophyly of particular clades, and it was suggested that 23% of animal species are polyphyletic when relying in their mtDNA data (Meyer & Paulay, Reference Meyer and Paulay2005; Meier et al., Reference Meier, Shiyang, Vaidya and Ng2006). It was also pointed out that one of the major flaws of the ‘barcode’ proposal is the use of a single molecular region, the 5′ extremity of cox1, and the question is whether this fragment is sufficient to achieve the purpose of diagnosing animal species (DeSalle et al., Reference DeSalle, Egan and Siddall2005). Alternately, other mitochondrial regions were shown to be as, or more, informative than the proposed ‘barcode’ region (Roe & Sperling, Reference Roe and Sperling2007; Wahlberg & Wheat, Reference Wahlberg and Wheat2008). By our own results on T. licus DNA polymorphism, it was possible to infer that the 5′ end of cox1 was the less variable of the mtDNA regions analyzed (fig. 2).
By using this region, part of the samples from TS_TM (clade C2 in fig. 4) was more closely related to T. l. laura (clade B1 in fig. 4) than to other individuals from TS_TM (clade C1 in fig. 4). Both adults from PV_RO are also within clade C2, and we confidently know they are morphologically compatible with the name T. licus albomaculata, as earlier discussed. In this way, we could alternatively name only the sample set in clade C2 in fig. 4 as T. licus albomaculata. The remaining samples from TS_MT might be considered as a different taxonomic unit from both T. l. laura and T. l. albomaculata. Considering just the less informative region of the 5′ end of cox1, we can conceive that there are two taxonomic entities in Mato Grosso, as presented by Lamas (Reference Lamas1995). For now, there is no data to fully discuss this issue, and additional sampling is undoubtedly necessary. However, it is possible to envisage that cryptic variation indeed exists within sympatric populations of T. licus subspecies, since morphologically divergent forms of adults of T. licus were never described in Tangará da Serra, MT (L.C. Almeida, personal communication). For other groups, such as thrips, it already has been shown that cryptic species can be collected even from the same individual host plant (Rugman-Jones et al., Reference Rugman-Jones, Hoddle and Stouthamer2010), reinforcing that morphological similarity is not indicative of genetic unity.
The origin of the population from São Paulo
The genetic distance and paired F ST data (table 3), coupled with the position of the samples from LI_SP grouped with samples from CO_AL and SM_AL in the genealogical network (fig. 3 I) and in the NJ topology (fig. 3 II), suggested that the population collected from the state of São Paulo must have a common origin with these populations from northeast Brazil. These data corroborate the hypothesis that ornamental plants infested with larvae of T. licus licus might have been transported from the northeast region of Brazil to the state of São Paulo (Almeida et al., Reference Almeida, Dias Filho and Arrigoni2007), giving origin to founders of the population currently attacking sugarcane plants. As this episode was recently reported (Almeida et al., Reference Almeida, Dias Filho and Arrigoni2007), there was not enough time to establish a marked genetic differentiation among these populations.
Conclusions
Castniidae is a very interesting moth family for several reasons: the day-flying or crepuscular habit, the resemblance to butterflies, and the economic importance of some of its species. Despite these, the group is poorly represented in collections (Miller, Reference Miller1986) and nearly nothing is known on its molecular systematic. The T. licus species complex is the classic example of a group where traditional morphological traits are not sufficient to disentangle complicated taxonomic relationships and where molecular data can be successfully employed (as found for other groups of butterflies (Silva-Brandão et al., Reference Silva-Brandão, Lyra and Freitas2009)). Speciation is not always accompanied by morphological change (Bickford et al., Reference Bickford, Lohman, Sodhi, Ng, Meier, Winker, Ingram and Das2007), and the recognition of cryptic entities must be made using alternative data as it is of utmost importance when pest/disease control is under discussion, as a failure in the correct identification of biological important species can be disastrous (Ellis et al., Reference Ellis, Blackshaw, Parker, Hicks and Knight2009; Paredes-Esquivel et al., Reference Paredes-Esquivel, Donnelly, Harbach and Townson2009; de León & Nadler, Reference de León and Nadler2010).
Our study on the genetic variability of T. licus populations from Brazil was the first attempt to relate the geographic range of this group to its genetic variation in order to determine the origin of a population and to delimit the taxonomy of the known subspecies. Sampling from other localities within Brazil, and abroad, would help to reveal the limits of the subspecies proposed for T. licus and to explore the mechanisms of speciation for this and other Neotropical species of castniids.
Acknowledgements
We thank Marcelo Duarte, for making available the Castniinae collection of Museu de Zoologia of Universidade de São Paulo (MZUSP), and Olaf H.H. Mielke and Mirna M. Casagrande, for gently allowing the visit to the collection deposited in the Dept. of Zoology, of Universidade Federal do Paraná (OM). We are also in debt to Sinval Silveira Neto and Roberto Antonio Zucchi for helping in the identification of specimens, and André V. L. Freitas for helping with adult traits descriptions. This research was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (projects 480619/2008-5 and 578509/2008-3), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES (PRODOC Process 0103/08-0) and FAPESP (Process 06/05365-3).