



Introduction
Muscular dystrophy is a group of genetic disorders characterised by degeneration of muscles. Different forms of muscular dystrophy present with a different degree of severity, the age of onset and location of muscle weakening (Ref. Reference Shieh1). There are numerous different muscular dystrophy variants; they result from various mutations in different genes (Ref. Reference Shieh1). Consequences of mutations can vary. For example, a complete loss of dystrophin as a result of out-of-frame deletion mutations in the DMD gene leads to a severe and rapidly progressive muscular dystrophy called Duchenne muscular dystrophy (DMD), while in-frame deletions, which do not disrupt the reading frame, lead to an internally truncated dystrophin production, leading predominantly to milder Becker muscular dystrophy (BMD) (Ref. Reference Beroud2). There is no cure for any form of muscular dystrophy, but treatment can help ease symptoms, delay disease progression and lead to a better quality of life in some forms of muscular dystrophy (Ref. Reference Mercuri and Muntoni3). Although recent advances allow delaying the progression of muscle degeneration and prolonging the longevity of life in patients by the use of medication (e.g. corticosteroid for DMD patients) and management of complications (Ref. Reference Yiu and Kornberg4), there has been much hope for the clinical application of new therapeutic approaches that can correct the genetic defects.
Among the muscular dystrophies, some common disorders show the potential to be treated by exon-skipping therapy, including but not limited to DMD, BMD, dysferlinopathy (DYSF), limb-girdle muscular dystrophy type 2C (LGMD2C) and merosin-deficient congenital muscular dystrophy type 1A (MDC1A) (Refs Reference Wyatt5–Reference Aoki8). In antisense-mediated exon-skipping therapy, pre-mRNA is targeted by synthetic antisense oligonucleotides (AOs), which hybridise to target sequences and alter the splicing. Hybridisation of synthetic AOs can block the splicing signal, and cause the splicing machinery of the cell to miss the exon and cause it to be spliced with its flanking introns (Ref. Reference Aartsma-Rus9). Skipping of exons using AOs allow synthesis of internally truncated but functional proteins as demonstrated numerous times in in vitro studies (Refs Reference Aartsma-Rus9–Reference Lu12), and in vivo animal model studies (Ref. Reference Goyenvalle13). There are many sites in the gene that can be targeted, including exonic splicing enhancer (ESE) (Ref. Reference Watakabe14), 5′ donor and 3′ acceptor splice sites (Ref. Reference Mann15). ESE is a preferred target sequence as it produces more consistent skipping of the exon (Ref. Reference Annemieke Aartsma-Rus16). Multiple exons can be skipped in the pre-mRNA by using a cocktail of AOs that target multiple exons (Ref. Reference Echigoya17). The efficacy of antisense-mediated exon skipping can be maximised by careful optimisation of which exons to skip, which sequences to target, which AOs to use and which injection methods, frequency and dosage to use (Refs Reference Goyenvalle13, Reference Echigoya17, Reference Echigoya18). Exon-skipping therapy is being explored as a potential tool for tackling genetic diseases (Refs Reference Yiu and Kornberg4, Reference Goyenvalle13), and especially appealing as it can correct genetic defects at the RNA level, instead of treating the symptoms (Fig. 1). However, it is worth noting that restoring the open reading frame (ORF) does not necessarily restore the function of proteins, as skipping of essential domains will disrupt protein function as in some DMD cases (Ref. Reference Bladen19).
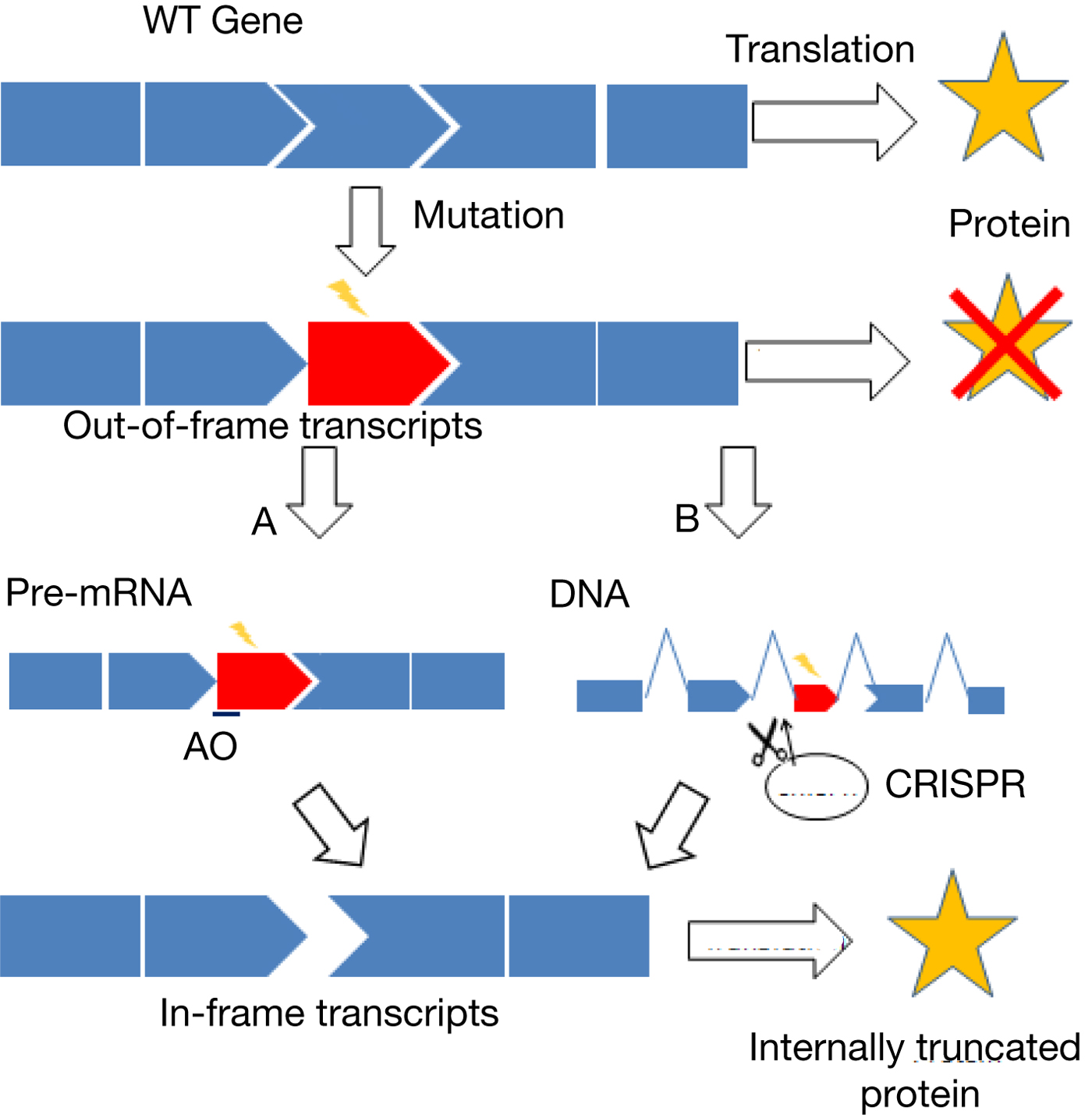
Fig. 1. Overview of exon skipping. Mutations can often lead to the production of a defective protein or no protein at all. Skipping of the defective exon and/or nearby exons while maintaining the ORF can lead to the production of an internally truncated protein. Examples of mutations that are potentially amenable to exon-skipping therapy include non-sense mutation, missense mutation, frameshift mutation and exon duplication and deletion (Refs Reference Lee47, Reference Yokota128–Reference Aoki130). (a) Antisense-mediated exon skipping. Antisense oligonucleotides (AOs) will target the mutated exon and/or nearby exons to induce the skipping of exons during splicing of pre-mRNA. (b) CRISPR-mediated exon skipping. CRISPR system will create mutations in the splice sites of the genome and cause permanent skipping of exons.
Clinical trials have been performed with eteplirsen (Exondys 51) developed by Sarepta Therapeutics to treat DMD, and has received accelerated approval from the US Food and Drug Administration (FDA) in September of 2016 (Ref. Reference Shimizu-Motohashi20). Clinical trials demonstrated that the progression of the disease was slightly slowed in treated patients, but showing limited clinical evidence for the efficacy of the drug (Ref. Reference Stein21). Nonetheless, the promising concept of restoring protein synthesis from the patient's own mutated genes by skipping exons holds great value in treating genetic disease, and recent studies on next-generation AOs that can enhance the therapeutic efficacy have shown positive results (Refs Reference Goyenvalle13, Reference Echigoya17) (Fig. 2).
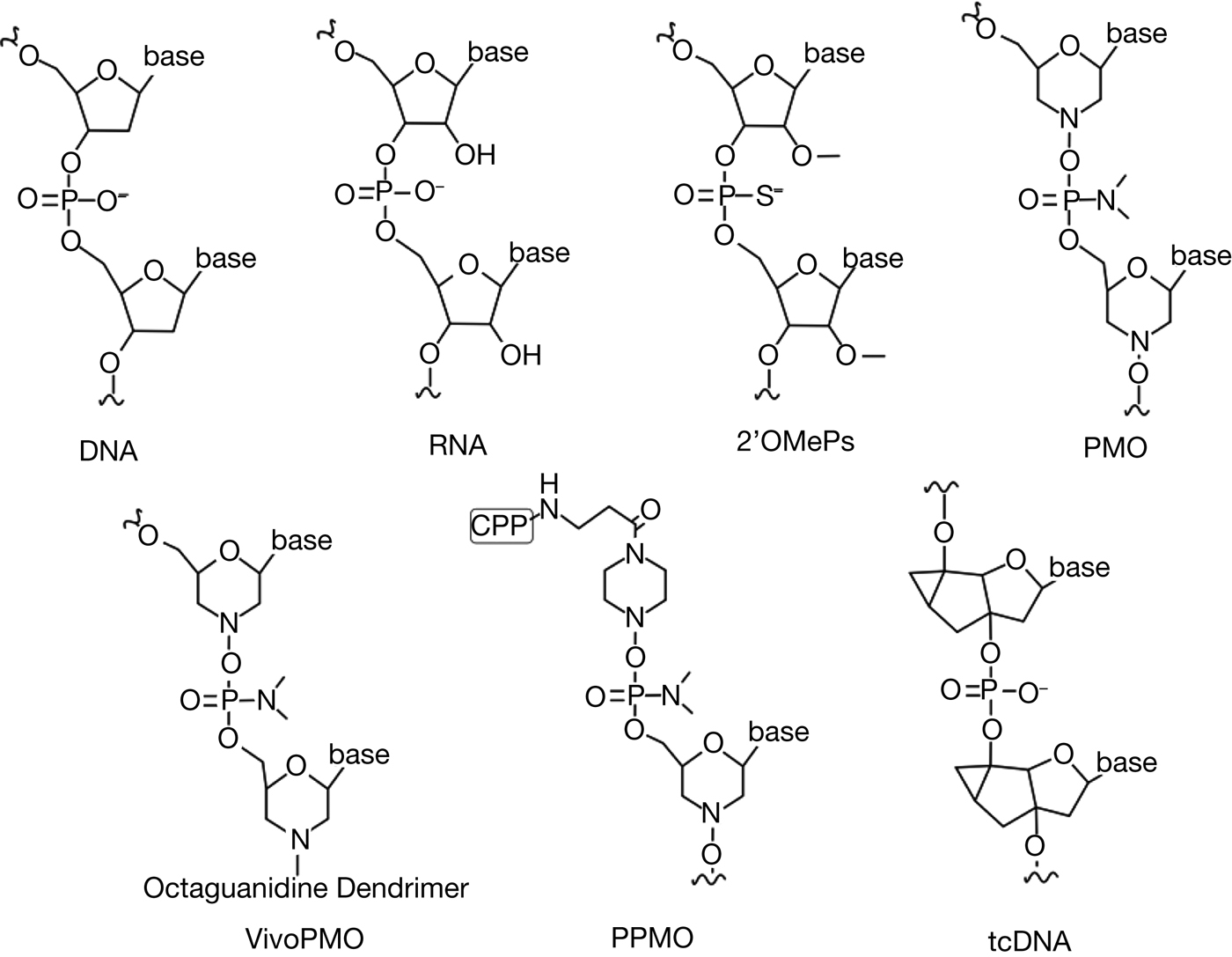
Fig. 2. Antisense oligonucleotides chemistry. In antisense-mediated exon skipping, various modifications are made in the nucleic acid that can confer therapeutic advantages. Modifications can be made in the backbone and/or the sugar ring.
Recently developing the alternative type of exon-skipping therapy adopts genome editing mediated by Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR associated (Cas) protein (Ref. Reference Long22). Cas9 introduces small insertion or deletion mutations (indels) that disrupt a splice site, leading to exon skipping (Ref. Reference Long22). On the other hand, the third-generation base editor (BE3) introduces a single base change in splicing regulatory sites to induce exon skipping (Refs Reference Gapinske23, Reference Mou24). Choosing an appropriate vector is a crucial step that impacts the safety and efficacy of CRISPR-based exon skipping. Guide RNAs should be also designed carefully, so that non-specific cleavage at other places in the genome does not occur. Exon skipping through CRISPR was demonstrated in human cells and animal models, confirming the rescued expression of desired proteins (Refs Reference Long22, Reference Gapinske23, Reference Amoasii25).
In this article, we will discuss advances in both antisense-mediated and CRISPR-mediated exon skipping, and discuss the types of muscular dystrophy that are treatable by exon-skipping therapy.
Application of exon skipping for various muscular dystrophies
Various forms of muscular dystrophies can each have a devastating impact on patients. Exon skipping has been studied to treat DMD from the early 1990s (Ref. Reference Pramono26). More recently, exon skipping has shown preliminary success in treating several other forms of muscular dystrophy, including LGMD2B, LGMD2C and MDC1A in animal models (Refs Reference Wyatt5, Reference Aoki8, Reference Malcher27). Identification of redundant exons in a gene is required prior to implementing exon-skipping therapies for each muscular dystrophy. A considerable amount of effort has been put into finding skippable exons with therapeutic benefit, but each gene has to be further explored and identification of more skippable exon targets are desired (Table 1).
Table 1. Partial list of therapeutic exon-skipping targets that are identified in DMD, DYSF, SGCG and LAMA2 genes

DMD and BMD
DMD is the most common type of muscular dystrophy that affects one in 5500 male births (Ref. Reference Mendell28). DMD patients begin to experience symptoms between 1 and 6 years of age accompanied by severe muscle degeneration. They mostly lose ambulation before the age of 12, and many die in their late teens if they are not medically intervened (Ref. Reference Eagle29). BMD presents with milder symptoms with a wide range of severity (Ref. Reference Beggs30). Both DMD and BMD are caused by a mutation in the DMD gene, which codes for the dystrophin protein. Clinical severity depends heavily on whether the mutation destroys the reading frame as in many DMD patients or maintains the reading frame as in most BMD patients (Ref. Reference Monaco31). The goal of exon-skipping therapy for DMD is to restore the reading frame by skipping the defective exon(s) and/or its nearby exon(s), to change the severe symptoms to the milder ones as seen in BMD (Ref. Reference Beroud2). DMD mutations cluster in a few hot spot regions, a major one being exon 45–55 and a minor one in exon 2–20 (Ref. Reference Juan-Mateu32). An FDA-approved AO drug eteplirsen targets exon 51 of the DMD gene, which is applicable for approximately 14% of DMD patients (Ref. 33). In addition, safety and dose-finding study of NS-065/NCNP-01 (also known as viltolarsen) targeting DMD exon 53 demonstrated significant increases in dystrophin protein, from 0.8 to 17.6% (immunostaining) and from 0 to 8.1% (western blotting) of normal levels in one patient, respectively (Ref. Reference Komaki34). Multi-exon skipping of exons 45–55 is also being studied, as patients harbouring deletions of exons 45–55 present with milder symptoms compared with patients with shorter in-frame exonic deletions (Ref. Reference Nakamura35), making it an ideal target for therapeutic exon skipping. In animal models and patient cells, successful multi-exon skipping (exons 45–55) has been demonstrated (Refs Reference Echigoya36–Reference Aoki38), increasing the applicability of the exon-skipping therapy to 47% of all DMD patients (Refs Reference Juan-Mateu32, Reference Nakamura35).
DYSF
DYSF is a rare muscular disease caused by a mutation in the dysferlin (DYSF) gene with the prevalence of 1: 100 000–200 000 (Ref. Reference Moore39). Dysferlinopathies consist of two distinct phenotypes: LGM2B and MM. Although LGM2B patients show muscle degeneration in proximal muscles of the limb girdle, MM patients present with symptoms in the distal muscles of the limb girdle and additionally show distal myopathy with anterior tibial onset which manifests in the distal and anterior compartments (Ref. Reference Illa40). However, phenotypes often become indistinguishable as both proximal and distal muscle function is lost in limb muscles (Ref. Reference Nguyen41). No drug has been approved for DYSF, and only palliative care is available. Dysferlin is thought to be involved in membrane repair and vesicle formation, and is present in various cells but predominantly in muscle cells (Ref. Reference Vandre42). Dysferlin consists of seven C2 domains, three ferlin domains, two dysferlin domains and one transmembrane domain. Many studies seem to suggest modular dispensability of dysferlin protein, as dysferlin, when deficient of certain C2 domains, still retains its membrane repair function (Ref. Reference Llanga43). Exon skipping is particularly anticipated to be a useful approach in treating DYSF as some patients with internally truncated dysferlin are reported to be only mildly affected (Ref. Reference Krahn44), suggesting the potential of skipping multiple exons in human to produce a therapeutic benefit. Naturally occurring mini-dysferlin molecules give us insights into potential targets for exon-skipping therapy (Ref. Reference Krahn45), and many mini-dysferlin plasmids have been constructed with promising results (Ref. Reference Llanga43). In a mouse model, exon 37 and 38 have been skipped but no functional study has been shown in vivo (Ref. Reference Malcher27). In addition, successful skipping of exon 32 was shown in patient myoblasts where membrane resealing function and membrane localisation of rescued dysferlin were observed (Ref. Reference Barthelemy46), which is applicable for 4% of DYSF patients (Ref. Reference Aartsma-Rus6). Our group also demonstrated successful skipping of exon 28–29, and showed restoration of membrane resealing ability in patient fibroblasts (Ref. Reference Lee47).
LGMD2C
LGMD2C is an autosomal recessive type of LGMD characterised by a childhood onset of progressive shoulder and pelvic girdle muscle wasting and atrophy that may also lead to a decline in cardiac and respiratory function (Ref. Reference Fayssoil48). It is caused by a mutation in the SGCG gene which codes for dystrophin-associated type II transmembrane protein, γ-sarcoglycan. There is currently no approved curative therapy, and treatment mainly focuses on supportive care (Ref. Reference Wyatt5). The most common SGCG mutation is a deletion of single thymine in a string of five thymine nucleotides in exon 6. Fixing the deletion of thymine mutation requires skipping of exons 4, 5, 6 and 7, which was shown to produce a functional protein encoded by exons 2, 3 and 8, named ‘Mini-Gamma’ in Drosophila (Ref. Reference Gao49). Exon skipping was tested in patient fibroblasts and myotubes using vivo-PMO, a PMO conjugated with octa-guanidine dendrimer cell-penetrating moiety, and demonstrated induction of correct exon-skipped mRNA products and production of Mini-Gamma proteins resulting in increased membrane integrity (Ref. Reference Wyatt5).
MDC1A
MDC1A, also known as congenital muscular dystrophy with laminin-α2 chain deficiency, is a severe type of autosomal recessive muscular dystrophy with the prevalence of one in 1 000 000 (Ref. Reference Durbeej50). Children affected by the disease show the generalised weakness of muscles, and delayed motor milestones. Most children affected never acquire ambulation and 30% of them die within the first decade of life because of respiratory tract infection (Ref. Reference Durbeej50). Mutations occur in the LAMA2 gene which codes for the laminin-α2 chain, a part of laminin-211 heterotrimer (Ref. Reference Durbeej50). Laminin-211 is a major component of the skeletal muscle basement membrane, which plays an important role in the functional integrity and maintenance of skeletal muscles (Ref. Reference Sanes51). Exon 4 of the LAMA2 gene has been skipped both in fibroblasts and mice using PMO, showing successful skipping and slightly increased survival of mice (Ref. Reference Aoki8). PMO uptake is possibly increased during myotube formation and in diseases like congenital muscular dystrophy where active muscle regeneration is prevalent, PMO treatment is anticipated to be more effective (Ref. Reference Aoki8).
Antisense-mediated exon skipping
AOs are short, synthetic nucleic acids with a modified backbone that bind complementary RNA by Watson–Crick base pairing (Ref. Reference Le Doan, Chavany and Helene52). Advantages of AOs include RNase resistance, high binding affinity to RNA, low-toxicity and efficient delivery to target cells (Ref. Reference Bennett53). Recent advances in synthetic nucleic acids allow numerous options of AOs that we can choose from, each with its advantages and disadvantages. Among them, there are a few AOs more commonly used that have yielded promising preclinical and clinical results for muscular dystrophy (Ref. Reference Sardone54).
2′ O-Methyl-phosphorothioate (2OMePS) is one of the second-generation AONs that is used for exon-skipping therapy. 2OMePS has ribose molecules with 2′ O-methyl modification and phosphorothioate modified negatively charged backbone, providing them with resistance to RNase degradation and allowing increased uptake. However, 2OMePS is cytotoxic and can cause liver damage and immune reaction from the host (Refs Reference Manoharan55, Reference Branda56). 2OMePS AO was used in an antisense-mediated exon-skipping drug drisapersen, developed for DMD patients. Drisapersen yielded a slight increase in dystrophin expression in treated patients but was rejected by the FDA after showing no statistically significant improvement in patients in a phase III trial (Ref. Reference Voit57). Injection site reaction and subclinical proteinuria were also reported during the trial (Ref. Reference Voit57).
Phosphorodiamidate morpholino oligomer (PMO) is an AO with an uncharged backbone of phosphorodiamidate linkages and morpholino moiety instead of the ribose sugar. Neutrally charged PMO does not readily interact with proteins, which contribute to its stability and safety making it suitable for clinical use (Ref. Reference Summerton58). However, one of the major challenges with using PMO for exon-skipping therapy is that it does not move across the cell membrane easily, especially in the heart, and the mechanism of its delivery to the cells are poorly understood (Ref. Reference Yokota59). Problems seem alleviated in DMD and MDC1A, where the changes in the cell membrane permeability are observed in regenerating myofibres (Refs Reference Aoki8, Reference McArdle, Edwards and Jackson60). Another hypothesis, called the leaky membrane hypothesis, is that AOs leak across the dystrophic muscle membrane, possibly facilitating entry of PMO (Refs Reference Aoki8, Reference McArdle, Edwards and Jackson60–Reference Ozawa, Hagiwara and Yoshida63). Also, a period of more active PMO intake is observed during myotube formation in regenerating fibres (Refs Reference Aoki8, Reference Novak64). Eteplirsen, the FDA-approved-drug for DMD, is an antisense PMO to skip exon 51 in DMD patients. Golodirsen (Sarepta Therapeutics) and NS-065/NCNP-01 or viltolarsen (NS Pharma) are PMO-based exon-skipping drugs that are currently under clinical trials, targeting exon 53 of the DMD gene (Refs Reference Komaki34, Reference Carver65). Although the FDA approval of the exon-skipping drug brings excitements to researchers, the limitation of PMO still persists and its limited efficacy needs to be addressed.
To overcome the limitations, PMOs are conjugated to different types of synthetic molecules. Peptide-conjugated PMOs (PPMO) are made by attaching positively charged, arginine-rich cell-penetrating peptides (CPP) to PMOs. The efficiency, toxicity and specificity of PPMO may differ depending on the CPP that is conjugated to the PMO (Refs Reference Copolovici66, Reference O'Donovan67). CPP aids in the transmembrane transfer of AO primarily via endocytosis (Ref. Reference Copolovici66), and sometimes through interaction with the negatively charged phospholipid bilayer (Ref. Reference Koren and Torchilin68). Efficient intake was observed in vivo with PPMO, producing internally truncated dystrophin and rescuing physiological abnormalities in animal models, including cardiac abnormalities (Ref. Reference Echigoya17). In one study of PPMO exon skipping for dystrophic dogs by Echigoya et al., little to no immune reaction was detected after systemic injection of PPMO (12 mg/kg for four injections or single injection, peptide sequence (RXRRBR)2XB, B = β-alanine; R = L-arginine; X = 6-aminohexanoic acid), and no obvious renal or hepatic damage was observed (Ref. Reference Echigoya17). However, another study that injected the same PPMO (60 mg/kg) into mice showed that three of the ten mice died of toxicity (Ref. Reference Wu69). Because CPP is non-specific, the toxic effect may be present in the liver and kidney and pose challenges for clinical use. Also, peptides may serve as antigens and cause an immune reaction, which would be hugely problematic considering the life-long treatment required for exon-skipping therapy. Understanding of CPP and its mechanism is still insufficient, and prediction of the efficacy and toxicity is still lacking (Ref. Reference Ye70).
Vivo-PMO is another conjugate of PMO that has octaguanidium dendrimer on triazine core attached to PMO. Octa-guanidine dendrimer is a non-peptide and non-natural conjugate characterised by its cost-effectiveness, convenient synthesis and effective cell penetration (Ref. Reference Morcos, Li and Jiang71). Vivo-PMO was utilised in an in vitro study where effective multi-exon skipping of the SGCG gene mRNA is shown, leading to expression of functional Mini-Gamma protein (Ref. Reference Durbeej50). The efficacy is similar to PPMO, however high toxicity was reported in one study that showed a high mortality rate in mice when injected at 11 mg/kg (Ref. Reference Ferguson, Dangott and Lightfoot72). The toxicity was hypothesised to be caused by self-hybridisation among vivo-PMOs leading to a cluster of positively charged guanidiums, which may have caused a change in red blood cell membrane structure and increased blood viscosity, resulting in blood ischaemia (Ref. Reference Ferguson, Dangott and Lightfoot72). Careful selection of vivo-PMO and addition of physiological saline are suggested as solutions to the coagulation vivo-PMO may cause. However, other experiments report no fatalities at 11 mg/kg of vivoPMO (Ref. Reference Aoki38).
Tricyclo-DNA (tcDNA) is another type of AO that contains three additional carbon atoms between C5′ and C3′ (Ref. Reference Renneberg and Leumann73). The exact mechanism of its uptake is unknown, but tcDNA is found to form nanoparticles in a physiological environment which may explain its effectiveness in cellular uptake (Ref. Reference Goyenvalle13). Quite notably, tcDNA shows great efficacy in inducing exon skipping in the heart, diaphragm and central nervous system (CNS), demonstrating 25–50 and 40% production of dystrophin compared with wild type in the diaphragm and heart, respectively, and displaying functional rescue of CNS functions in mice (Refs Reference Goyenvalle13, Reference Relizani74). tcDNA has a strong binding affinity towards RNA, allowing the use of shorter nucleotides for therapeutic benefit (Ref. Reference Renneberg75). Shorter nucleotides allow reducing the mass needed to induce exon skipping, reducing the toxic effect that may happen. One study shows a sign of mild nephrotoxicity, characteristic of phosphorothioate backbone accumulation, and moderate inflammation in the liver (Ref. Reference Relizani74). However, more studies in toxicology in different animals are needed to predict its effect on the human.
Sequence optimisation
Each AO used for exon-skipping therapy has a specific target sequence. ESE sites are usually targeted by AO-mediated exon-skipping therapy, but even within the ESE sites, differences in efficacy may go up to tenfold (Ref. Reference Echigoya18). In order to maximise the efficacy in exon-skipping therapy, it is crucial to optimise the sequence to the best of our ability. Many studies have taken a retrospective approach that tests numerous target sequences to find the best target sequence (Ref. Reference Aartsma-Rus76); however, a predictive approach that attempts to analyse various parameters affecting the efficacy and finding an optimal sequence in silico is emerging (Ref. Reference Echigoya18). Among the many parameters, some factors allow better prediction while some have less influence. One of the biggest factors that affect exon-skipping efficacy is the difference in free energy (dG) between bound state and unbound state of AOs to the exon, in other words binding energy of the oligo to the target sequence. Percentage of GC content, the length and oligonucleotide dimerisation all affect the binding energy (Ref. Reference Echigoya18). Another major factor that determines the efficacy of exon skipping is the distance of the target site from the acceptor site measured as Average Cumulative Position (ACP) (Ref. Reference Pramono77). In the study done by Pramono et al., 23 AOs were tested for exon-skipping potential, and all AOs that had their first nucleotide within the first third of exon (closer to the acceptor site) showed efficient exon skipping, while all AOs that had their first nucleotide in the middle and last third of exon show inefficient or non-efficacious exon skipping (Ref. Reference Pramono77). This may be because of the cotranscriptional nature of splicing, where AOs need to compete with the splicing apparatus as mRNA is being transcribed, requiring the AO to bind as early as possible to exposed exonic sequences. By creating a formula that incorporates dG, ACP and Malueka type, a study by Echigoya et al. predicts efficient versus inefficient PMOs in silico with 89% success rate (Ref. Reference Echigoya18).
Malueka et al. in their study describe different types of exons and group them in five different groups based on their splicing mechanism (Ref. Reference Malueka78). Different types of exons have different exon-skipping potential. Accessibility of the target site is another factor that influences the exon-skipping efficacy. The open conformation of mRNA transcript allows access to the target sequence by exon-skipping AOs; therefore, attempts were made to calculate accessibility for each base of the target exon (Ref. Reference Echigoya18). A thermodynamic model of cotranscriptional pre-mRNA folding was used to calculate L3 value which is the average engagement of the target bases during transcription (Ref. Reference Wee79). A low L3 value indicates that the target bases are more accessible to AOs during transcription, meaning more potential for effective exon skipping. Furthermore, the binding propensity of splicing factors are estimated by Neighbourhood Inference (NI) scoring (Ref. Reference Stadler80), and used to estimate exon-skipping efficacy.
When optimizing the sequences of AOs for exon skipping, not only the efficacy but also toxicity has to be considered. Self-dimerisation of oligos can affect the efficacy and also cause toxicity as demonstrated by injection of low-dose vivo-PMO causing fatalities in mice when there was dimerisation within the AOs (Ref. Reference Ferguson, Dangott and Lightfoot72). Sequences of AOs that may hybridise with off-target sequences may cause unwanted side effects, including apoptosis (Ref. Reference Stein81).
Injection methods
Depending on the injection location, dose and frequency, the efficacy in exon skipping may vary. Although the ideal injection methods may be different for each type of AO or disease, there are some data that can work as a guideline. Intramuscular injection delivers AOs to a specific muscle group, allowing efficient delivery of AOs to local muscles (Ref. Reference Aoki38). It can be used to assess exon skipping in the specific muscle group. Intrathecal injection can be used to deliver AOs to the CNS, as demonstrated in SMA patients (Ref. Reference Hache82). Intraperitoneal injection reported promising exon-skipping potential, and it shows effective delivery of AOs to the diaphragm and abdominal muscles in mice (Ref. Reference Wu83). Subcutaneous injection (SC) has been applied for the clinical trial of drisapersen (Ref. Reference Goemans84); however, SC injection is known to cause specific local skin reactions originating around the injection site (Ref. Reference van Meer85). Intravenous (IV) injection is of the most promising injection methods used by the researchers for muscular dystrophy. One of the advantages of IV injection is that higher volume can be delivered via IV injection compared with other methods (Ref. Reference Riebesehl86).
CRISPR-mediated exon skipping
CRISPR-mediated genome editing is a recent technology that is rapidly developing and has a huge potential in genetic therapies. CRISPR system shows a great flexibility in genetic modification but in vivo delivery of CRISPR-Cas9 and non-specific off-target effects remain challenging (Ref. Reference Roy87). There are many ways CRISPR can be used to treat genetic diseases including homology-directed repair (HDR)-mediated gene correction, single base pair correction, exon deletions and frame correction via insertion and deletion (INDEL) mutations (Refs Reference Long22, Reference Gapinske23, Reference Bengtsson88). In this review, CRISPR-mediated exon skipping will be discussed where INDEL mutations and single base pair changes are created in sites crucial for splicing to induce skipping of exons. Crucial splice sites include ESE sites (Ref. Reference Amoasii25), donor sites and acceptor sites (Ref. Reference Long22). Induction of exon skipping is advantageous compared with other methods in that the donor template is not required and a single guide RNA (sgRNA) is sufficient to skip a target exon in the RNA (Fig. 3).
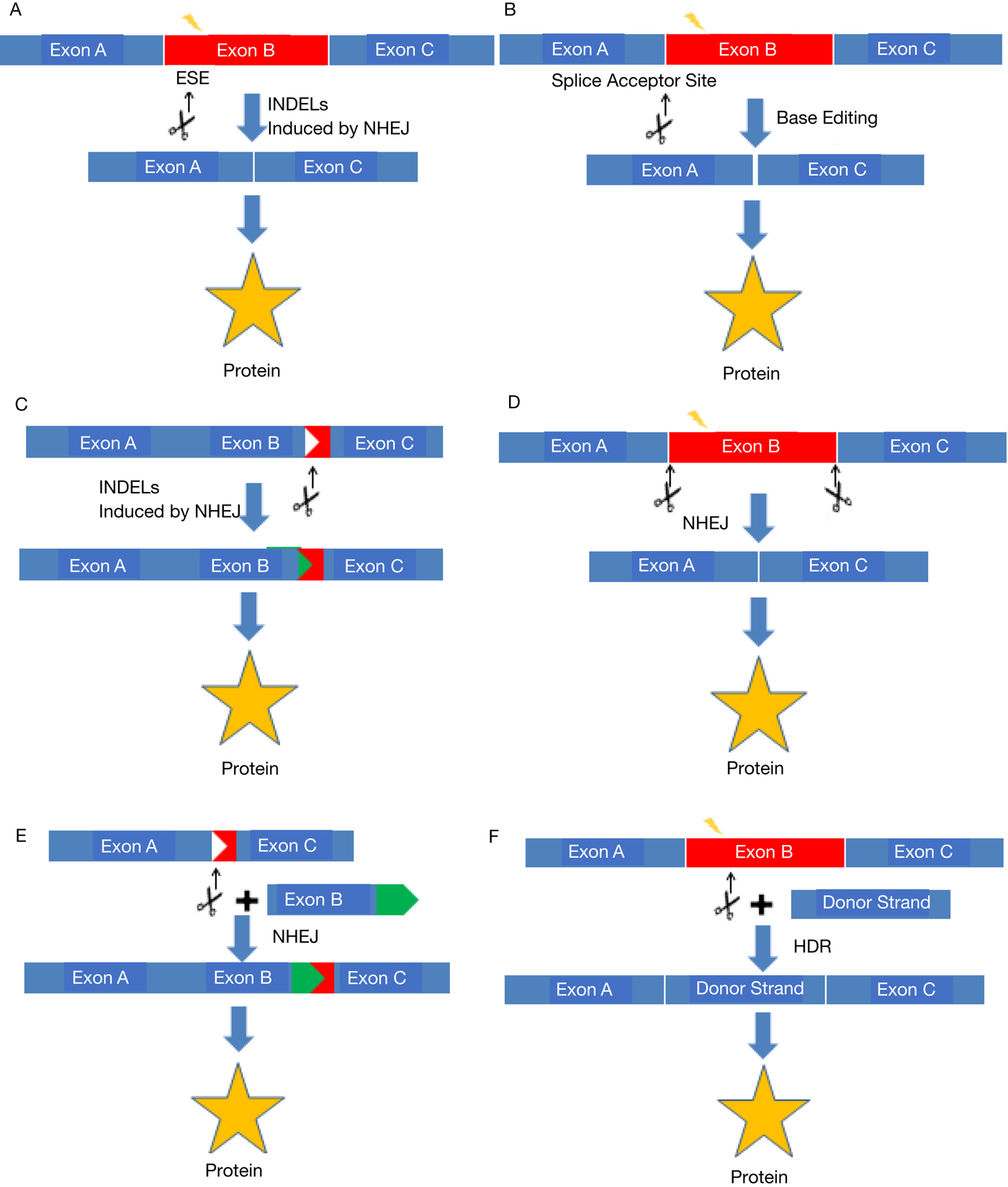
Fig. 3. Summary figure of different CRISPR-mediated genome editing methods. CRISPR can be used in various ways to rescue protein expression from a mutated gene. (a, b) Exon skipping. Splice site mutations are created by INDELs (a) or base editor (b) to induce exon skipping in the transcripts. (c) Reframing. For out-of-frame mutations, INDELs are created to correct the reading frame of the gene. (d) Exon deletion. Two double-stranded breaks flanking an exon(s) are created, inducing deletion of the exon(s). (e) Homology independent targeted integration (HITI). A double-stranded break in a gene and another double-stranded break in the donor template to induce insertion of a gene fragment. (f) Homology directed repair (HDR). Double-stranded break created in the gene is repaired by the donor template in the presence of homologous regions flanking the cleavage site.
Conventional CRISPR system works through CRISPR-associated proteins such as Cas9 endonuclease directed by the guide RNA, which makes a double-stranded break in target sequences (Ref. Reference Hale89). Double-stranded breaks are mostly repaired by non-homologous end joining (NHEJ) repair system, which can often create INDEL mutations. In CRISPR-Cas9-based exon skipping, Cas9 protein and guide RNAs targeting splicing regulatory sites are administered, creating INDEL mutations at the splice regulatory sites. However, the efficiency of exon skipping may be difficult to predict, as some sites when mutated induce exon skipping more readily than others, and different mutations from a sgRNA may or may not induce exon skipping (Refs Reference Long22, Reference Kapahnke, Banning and Tikkanen90). Unexpected deletions of multiple exons have also been observed from a splice site mutation induced by a sgRNA, but the mechanism is poorly understood (Ref. Reference Kapahnke, Banning and Tikkanen90); better characterisation of alternative splicing induced by INDEL mutations will allow the researchers to manipulate splicing efficiently (Refs Reference Mou24, Reference Kapahnke, Banning and Tikkanen90). CRISPR-mediated skipping of DMD exon 51 and many other exons was demonstrated by researchers in human cells, mouse models and a dog model of DMD (Refs Reference Long22, Reference Amoasii25, Reference Amoasii91, Reference Amoasii92).
Recently, the protein synthesised by the fusion of Cas9 protein with cytidine deaminase is shown to edit the target base without making a double-stranded break (Ref. Reference Komor93). Catalytically dead cas9 (dcas9) created by mutations allow sgRNA-mediated binding to the target sequence without its ability to cause a double-strand break, and fused cytidine deaminase acts on the base to change cytidine into uracil (Ref. Reference Komor93). Unlike NHEJ-mediated exon skipping which relies on stochastic mutations to induce exon skipping, BE3 can make a specific change of a selected base (Ref. Reference Gerhke, Cervantes, Clement, Pinello and Joung94). In one in vitro study, BE3 is utilised to induce exon skipping in human cells by switching the guanosine at the intron–exon boundary into adenosine, disrupting the highly conserved consensus sequence of the splicing acceptor site (Ref. Reference Gapinske23). The same study compares its efficacy with cas9 based exon skipping, and shows that BE3 demonstrates an equal or greater degree of exon skipping in tested exons (Ref. Reference Gapinske23). Further studies are being done to increase the precision of genome editing by testing Cas9 proteins with various mutations (Ref. Reference Gerhke, Cervantes, Clement, Pinello and Joung94).
CRISPR optimisation
CRISPR system is efficient in itself, but there are various ways it can be optimised for better functionality. When CRISPR is encoded in a plasmid, the selection of a promoter can affect the pattern of expression which may determine its effectiveness. Tissue specificity of certain promoters may be desirable for CRISPR application, as it allows reducing non-specific effects in non-target tissues. However, RNA polymerase III promoter (U6) is typically used for the expression of sgRNA to avoid extensive processing such as 5′ capping and 3′ poly-A tailing (Ref. Reference Ma95). Furthermore, the choice of Cas enzyme is another important factor to consider. Cas9 is a type II CRISPR enzyme that is most commonly used, and Cas12a (aka Cpf1) is a type V Cas enzyme that is also being used for exon skipping (Ref. Reference Swarts and Jinek96). Among the same type of Cas enzymes, enzymes from different species may exhibit different characteristics that confer advantages and disadvantages to each enzyme. Differences among the enzymes include their protospacer adjacent motif (PAM) recognition, cleavage characteristics, target specificity and the size of the enzymes (Refs Reference Kim97, Reference Friedland98).
Sequences of sgRNA have to be selected meticulously, as sgRNA nucleotide composition can affect its efficiency in loading, stability and mutagenic activity (Ref. Reference Moreno-Mateos99). Guanine-rich and adenine-depleted nucleotide composition and a guanine adjacent to the PAM are shown to correlate with high mutagenic activity of CRISPR-cas9 (Ref. Reference Moreno-Mateos99). Eight guanines are needed to form G-quadruplex, which is suggested to contribute to sgRNA stability (Ref. Reference Moreno-Mateos99). G-quadruplex structure, which is formed both on sgRNA and the double-stranded DNA, stabilises R-loops which are formed during CRISPR and DNA interaction, enhancing the mutagenic activity (Ref. Reference Moreno-Mateos99). sgRNAs with one or two mismatches or deletions in the 5′ end, denoted as alternative sgRNA, are shown to have similar mutagenic activity compared with canonical sgRNAs, and utilizing alternative sgRNAs would increase the number of potential target sites eightfold (Ref. Reference Moreno-Mateos99). Careful optimisation of sgRNA can lead to higher efficiency and reduced non-specific mutagenic activity. In addition, further characterisation of sgRNA sequences with a better-predicted efficacy and toxicity will facilitate the development of CRISPR-mediated exon skipping.
Vector selection
For CRISPR-mediated exon skipping, viral vectors, non-viral vectors or a combination of both may be used (Ref. Reference Yin100) (Table 2). Adeno-associated virus (AAV) is the most common type of viral vector used for the delivery of the CRISPR system in vivo. AAV is a virus of the parvoviridae family that requires a helper virus for its replication, and its advantages include its safety profile, specificity, diversity of target cells and efficient transduction (Ref. Reference Mingozzi and High101). Numerous clinical trials are done with AAV, and different serotypes of AAV present with their advantages for specific circumstances (Ref. Reference Mingozzi and High101). For example, certain capsids of the AAV serotypes can offer specific tissue tropism such as AAV8 that deliver genes to liver effectively (Refs Reference Kattenhorn102, Reference Grimm103), and certain serotypes such as AAV9 show the ability to cross the blood–brain barrier, making it ideal for the delivery of CRISPR system to the CNS (Ref. Reference Xie104). Aside from naturally discovered serotypes of AAV, rAAV vectors are continuously being engineered for improved performance (Ref. Reference Li105). However, challenges of AAV as a vector include relatively small transgene capacity, pre-existing humoral immunity against AAV and dose-dependent toxicity (Ref. Reference Asokan, Schaffer and Samulski106). Lentiviral vector is another potential therapeutic viral vector. Lentivirus is known to evade the host immune system very well (Ref. Reference Abordo-Adesida107), and show effective transduction of quiescent cells (Ref. Reference Naldini108). Lentivirus is also advantageous in that it has larger transgene capacity than AAV (~9 kb) allowing single vector delivery of both cas9 and gRNA. However, insertion of DNA may cause insertional mutagenesis, which may cause cancer (Ref. Reference Schambach109). Delivery of a gene via Lentivirus has succeeded many times in clinical trials and in vivo (Refs Reference Abordo-Adesida107, Reference Kuruvilla, Sasmita and Ling110, Reference Palfi111). Apart from viral vectors, there are non-viral means of CRISPR delivery in vivo. Although transfection may be less efficient than viral vectors, non-viral vectors are generally thought to be safer. Naked DNA, mRNA alone or with other physical means such as hydrodynamic delivery, gene gun, electroporation, sonoporation and magnetofection have been attempted (Refs Reference Mehier-Humbert and Guy112, Reference Yin113) but these methods showed low efficiency in human, and the half-life of naked plasmid DNA in the cytoplasm ranged from 50 min to 5 h (Ref. Reference Vaughan, DeGiulio and Dean114). Synthetic delivery vectors face many steps of barriers that affect the transfection efficiency such as association with the cell surface and nuclear barrier it has to cross. Selective accumulation at the target site is also difficult for most synthetic vectors. However, careful studying of each barrier and the vector is leading to advances and modifications of synthetic vectors to improve the efficiency. Modified Cas9 mRNA was delivered in mice via lipid nanoparticles, together termed nano-cas9, along with associated helper lipids and AAV vector carrying sgRNA, and induced HDR-mediated genome correction for 6% of hepatocytes in mice (Ref. Reference Yin100). The same nano-cas9 produced double-strand break in 77% of cells tested in vitro (Ref. Reference Yin100). Furthermore, ZAL nanoparticle is synthesised to contain chemical and structural roles of zwitterionic and cationic lipids, and successfully demonstrated co-delivery of Cas9 mRNA and sgRNA in mice causing CRISPR editing of liver, kidney and lung cells (Ref. Reference Miller115).
Table 2. Summary of vector choices for CRISPR delivery with its advantages and disadvantages

Challenges and future implications
In this paper, we discussed briefly both antisense-mediated and CRISPR-mediated exon-skipping therapy and discussed promising exon-skipping target sites in DMD, DYSF, SGCG and LAMA2. Although exon skipping shows promising results with recent advances, there are challenges that need to be overcome.
In many studies of exon-skipping therapy, one of the major hurdles is possible off-target effects (Refs Reference Wu69, Reference Ferguson, Dangott and Lightfoot72, Reference Mingozzi and High101). Furthermore, different tissues have different efficiency of exon skipping, with heart proving to be very difficult to induce exon skipping. Since cardiomyopathies are often fatal, AOs and vectors for CRISPR that can efficiently target the heart for exon skipping are crucial. In general, AOs and delivery vectors for CRISPR need to be better characterised of its toxicity and efficiency, as many AOs and vectors are sitting on the table and every bit of information about them will help the community select the best one and modify it to achieve the best possible results.
Even after the problem of toxicity and efficiency is resolved, exon skipping comes with the inherent limitation of applicability. Each exon-skipping drug can only treat a subset of patients while patients that carry a mutation in the essential domain of a protein are not treatable by exon-skipping therapy. Nonetheless, the identification of more and better exon-skipping targets is vital as identification of each skippable exon may allow numerous patients to be treated (Table 1). Multi-exon skipping can further resolve the issue as targeting several exons at the same time can increase the applicability to a larger population. However, developing exon skipping for multiple exons need to overcome the regulatory challenges (Ref. Reference Aslesh, Maruyama and Yokota116). Each AO targeting different exon must go through separate laborious and expensive FDA approval process, which becomes especially troublesome in multi-exon skipping using a cocktail of AOs since each AO does not necessarily have a therapeutic benefit in itself and can be rather toxic. With increasing prevalence and significance of sequence-specific drugs, novel regulatory measures are needed to speed up the development of AO and CRISPR-mediated exon-skipping therapies.
Sequence optimisation discussed above is also a critical step that we have to improve on for efficient exon skipping. A sequence optimisation formula was tested with PMOs for DMD, but it is unclear whether the same formula could accurately predict skipping efficiency in other genes with various other AOs (Ref. Reference Echigoya18). Furthermore, taking the cellular environment into account is currently challenging, and a much deeper understanding of protein and structure dynamics is required to compute environmental factors into efficiency prediction. CRISPR-mediated exon skipping also requires thorough screening efforts for appropriate sgRNAs. Many sgRNAs do not result in efficient cleavage, and certain sgRNAs can act on non-specific targets, potentially disrupting other genes. Mutations in each target site show a huge difference in exon-skipping efficiency, and different mutations created by the same sgRNA can also have varying consequences (Ref. Reference Mou24). Although large-scale screening efforts to determine optimal sgRNA sequences have been made and various factors were characterised, more techniques will be developed in the future (Refs Reference Moreno-Mateos99, Reference Doench117).
AO-mediated and CRISPR-mediated exon-skipping therapies each face its own barrier to overcome. AOs target pre-mRNA to cause a transient change in splicing to cause exon skipping (Ref. Reference Geary118). The transient nature of AO-mediated exon skipping means that the patients with genetic defects would have to be medicated for the rest of their lives. However, the current cost of AOs may not be affordable for many patients, with eteplirsen costing $300 000 per year (Ref. Reference Lim, Maruyama and Yokota119). Another problem faced by AO-mediated exon skipping is that the use of cocktail AOs requires FDA approval of each AO in the cocktail (Ref. Reference Aartsma-Rus120). Many mutations require skipping of multiple exons to have therapeutic benefit, and multiple exon skipping requires the use of a cocktail of AOs targeting different exons (Ref. Reference Echigoya36). On the other hand, CRISPR-mediated exon skipping presents with another set of challenges. CRISPR causes a permanent change in the genome (Ref. Reference Kyrychenko121). Permanent correction of a genetic defect may be desirable, but the irreversible nature may mean that if the therapy is not applied carefully enough, the patient may suffer from side effects permanently. Currently used vectors for the delivery of CRISPR express the enzyme for a prolonged period of time even after the desired gene-editing outcome is created (Ref. Reference Colella, Ronzitti and Mingozzi122). This persistence of CRISPR enzyme in the system can pose long-term risks, as the chance of off-target modification may increase as the enzyme stays in the system longer. Vectors and plasmids that promote transient expression of the protein would be able to reduce the safety risks.
The potential of exon-skipping therapy and its techniques is exciting, and in due time it will open up a new era of personalised medicine and genome editing. With improved efficacy, personalised medicine will be available for each patient, where specific mutations of each patient will be treated accordingly. Patient genome sequences will be analysed for possible non-specific binding sites, and the optimal number of exons will be skipped for maximal benefit. We believe that exon-skipping therapy and optimisation of such techniques will be the cornerstone of the new era that is coming.
Acknowledgement
This work was supported by the Friends of Garrett Cumming Research Chair Fund, the HM Toupin Neurological Science Research Chair Fund, the Muscular Dystrophy Canada, Canada Foundation for Innovation (CFI), Alberta Advanced Education and Technology (AET), Canadian Institutes of Health Research (CIHR), Jesse's Journey – The Foundation for Gene and Cell Therapy, Rare Disease Foundation/BC Children's Hospital Foundation, Heart & Stroke Foundation Alberta, the University of Alberta Faculty of Medicine and Dentistry, and the Women and Children's Health Research Institute (WCHRI).
Conflict of interest
None.





