


Introduction
The genus Prunus includes more than 200 species of flowering shrubs and trees in the Rosaceae family and has great economic importance, as it includes the cultivated almond, peach, plum, cherry and apricot (Ercisli, Reference Ercisli2004; Yilmaz et al., Reference Yilmaz, Ercisli, Asma, Dogan and Kafkas2009).
All commercially grown sweet cherries grafted or budded on rootstocks belong to different Prunus species. Sweet cherry rootstocks can directly influence productivity, precocity, tree size, tree architecture, fruit size and fruit quality of sweet cherry cultivars. The choice of certain rootstocks will also influence many horticultural decisions such as pruning, training, tree support and labour management (Jimenez et al., Reference Jimenez, Pinochet, Gogorcena, Betran and Moreno2007; Cantín et al., Reference Cantín, Pinochet, Gogorcena and Moreno2010; Radunic et al., Reference Radunic, Jazbec, Pecina, Cosic and Pavicic2011). Traditionally, sweet cherry rootstocks were identified based on morphological characteristics and those parameters were strongly affected by the environment and the developmental stage of plant as well. This dependence on morphological traits makes rootstock identification nearly impossible particularly during the dormant season of plants. Moreover, botanical classification of species within this genus is sometimes controversial, partly because of the easiness of interspecific hybridization of the Prunus genus (Dosba et al., Reference Dosba, Bernhard and Zanetto1994).
For breeding and commercialization of promising rootstock candidates, a precise determination and discrimination of these materials is requested. In the case of rootstocks, it is very difficult to observe their morphological traits after grafting. Therefore, markers independent from the environment are necessary for reliable identification and discrimination of accessions. Superiority of molecular markers over morphological characterization in fruit species is well established and widely accepted (Ercisli et al., Reference Ercisli, Agar, Orhan, Yildirim and Hizarci2007; Zamani et al., Reference Zamani, Sarkhosh, Fatahi and Ebadi2007; Kafkas et al., Reference Kafkas, Ozgen, Dogan, Ozcan, Ercisli and Serce2008; Duminil and Di Michele, Reference Duminil and Di Michele2009; Szikriszt et al., Reference Szikriszt, Hegedus and Halasz2011).
Microsatellites or simple sequence repeat (SSR) markers have been very useful for studying the extent and distribution of genetic variability in wild and cultivated plants including various Prunus species (Cheng and Huang, Reference Cheng and Huang2009; Guarino et al., Reference Guarino, Santoro, De Simone and Cipriani2009; Nas et al., Reference Nas, Bolek and Bardak2011) and the number of microsatellite loci available, in particular, for the Prunus genus has greatly increased. In Prunus, microsatellites have been used for germplasm characterization (Lacis et al., Reference Lacis, Rashal, Ruisa, Trajkovski and Iezzoni2009), determination of genetic diversity (Bouhadida et al., Reference Bouhadida, Martin, Eremin, Pinochet, Moreno and Gogorcena2007), germplasm management (Cheng and Huang, Reference Cheng and Huang2009), parentage analysis (Yamamoto et al., Reference Yamamoto, Mochida, Imai, Haji, Yaegaki, Yamaguchi, Matsuta, Ogiwara and Hayashi2003), cultivar identification (Xuan et al., Reference Xuan, Wang, Buchele, Moller and Hartmann2009) and mapping genetic linkage (Lalli et al., Reference Lalli, Abbott, Zhebentyayeva, Badenes, Damsteegt, Polak, Krska and Salava2008). In Prunus, microsatellites developed in one species have been used in a different species, demonstrating their transferability and ability to detect polymorphism (Wunsch, Reference Wunsch2009).
In cultivated sweet cherries, many reports related to SSR analysis have been published in different countries (Cheng and Huang, Reference Cheng and Huang2009; Lacis et al., Reference Lacis, Rashal, Ruisa, Trajkovski and Iezzoni2009; Gulen et al., Reference Gulen, Ipek, Ergin and Akcay2010). However, much less has been done to assess genetic diversity of wild cherry species by molecular markers.
The objective of this study was to select a set of microsatellite loci useful to detail polymorphism in different accessions belonging to Prunus species used as rootstocks for sweet cherry. It is expected that the information presented here would be useful for selection and more efficient utilization of this germplasm as rootstocks for sweet cherries in future.
Materials and methods
Plant material
For SSR and genetic relationship studies, a total of 184 accessions including representatives of the species Prunus avium (110 accessions), Prunus mahaleb (40 accessions), Prunus cerasus (29 accessions) and Prunus angustifolia (5 accessions) were used for SSR analysis. These accessions were previously selected from wild cherry populations in a national-wide sweet cherry rootstock selection study conducted in the north-western part of Turkey (supplementary material available online at http://journals.cambridge.org/). All accessions are maintained in a germplasm collection at the Black Sea Agricultural Research Center in Samsun, Turkey. Three standard Prunus rootstocks, SL64 (P. mahaleb L.), F12/1 (P. avium L.) and Montmorency (P. cerasus L.), were also included in the SSR analysis as a reference.
DNA extraction
Genomic DNA was extracted from young leaf tissue using the Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the instructions provided by the manufacturer. Subsequently, an RNase treatment was performed on the eluted DNA samples. Purity and concentration of the DNA were checked both on 1% (w/v) agarose gels and by using a NanoDrop® ND-1000 Spectrophotometer.
SSR analysis
We initially checked 16 SSR primers. These 16 SSR primers are widely used in Prunus for molecular characterization (Akpinar et al., Reference Akpinar, Kocal, Ergul, Kazan, Selli, Bakir, Aslantas, Kaymak and Saribas2010; Gulen et al., Reference Gulen, Ipek, Ergin and Akcay2010; Ercisli et al., Reference Ercisli, Agar, Yildirim, Duralija, Vokurka and Karlidag2011; Yilmaz et al., Reference Yilmaz, Paydas-Kargi, Dogan and Kafkas2012). However, ten of them gave better amplification in this study. Therefore, ten SSRs were selected to check polymorphism by capillary electrophoresis in 184 accessions of the four different Prunus species (Table 1). Polymerase chain reaction (PCR) was conducted in a volume of 10 μl and contained 15 ng genomic DNA, 5 pmol of each primer, 0.5 mM dNTP, 0.5 unit GoTaq DNA polymerase (Promega), 1.5 mM MgCl2 and 2 μl 5 × buffer. The forward primers were ‘labelled’ with WellRED fluorescent dyes D2 (black), D3 (green) and D4 (blue) (Pro-ligo, Paris, France). Reactions without DNA were included as negative controls. PCR amplification was performed using the Biometra® PCR System. The amplification conditions consisted of an initial denaturation step of 3 min at 94°C, followed by 35 cycles of 1 min at 94°C, 1 min at 52–56°C and 2 min at 72°C with a final extension at 72°C for 10 min. For the determination of polymorphisms, the PCR products were run on a CEQTM 8800 XL Capillary Genetic Analysis System (Beckman Coulter, Fullerton, CA, USA). The analyses were repeated at least twice to ensure reproducibility of the results. In each run, SL64, F12/1 and Montmorency were included as reference rootstocks.
Table 1 Simple sequence repeat primer pairs used in this study to characterize 184 rootstocks belonging to Prunus

Genetic analysis
The genetic analysis program ‘IDENTITY’ 1.0 (Wagner and Sefc, Reference Wagner and Sefc1999) was used according to Paetkau et al. (Reference Paetkau, Calvert, Stirling and Strobeck1995) for the calculation of number of alleles, expected and observed heterozygosity (H e and H o), estimated frequency of null alleles, and the probability of genetic identity per locus. Probability of genetic identity corresponds to the probability of two random individuals displaying the same accessions. That means if probability of identity (PI) = 1, it indicates that the primer pair is not able to distinguish between the accessions, and vice versa the smaller is the PI value, the more informative the SSR primer pair (locus) is. Null alleles are non-amplified alleles that, when segregating with another allele, result in an apparent homozygote. For microsatellites, such null alleles can arise when mutations occur in the flanking regions, preventing one or both of the primers from binding (Holm et al., Reference Holm, Loeschcke and Bendixen2001). Genetic similarity was determined by the program ‘MICROSAT’ (version 1.5) (Minch et al., Reference Minch, Ruiz-Linares, Goldstein, Feldman and Cavalli-Sforza1995) using the proportion of shared alleles, which was calculated by using ‘ps (option 1 − (ps))’, as described by Bowcock et al. (Reference Bowcock, Ruiz-Linares, Tomfohrde, Minch, Kidd and Cavalli-Sforza1994). The results were then converted to a similarity matrix, and a dendrogram was constructed with the unweighted pair-group method with arithmetic mean (UPGMA) method (Sneath and Sokal, Reference Sneath and Sokal1973) using software NTSYS-pc (Numerical Taxonomy and Multiware Analysis System, version 2.0) (Rohlf, Reference Rohlf1988).
Results
In the study, a total of 133 alleles with an average of 13.3 alleles per locus obtained. The number of alleles per primer ranged from 10 (UDAp-401, UCD-CH21 and CPSCTO10 primers) to 20 (UCD-CH31) (Table 2).
Table 2 List of genetic parameters obtained with simple sequence repeat used in this study
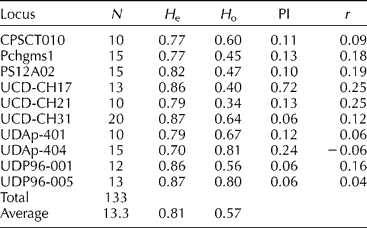
N, number of alleles; H o, observed heterozygosity; H e, expected heterozygosity; PI, probability of identity; r, null allele frequencies.
We observed an average SSR heterozygosity (H o) of 0.57, while the H e was 0.81. The H o, identified by each primer pair, ranged between 0.34 (UCD-CH21) and 0.81 (UDAp-404) and the H e was found between 0.70 (UDAp-404) and 0.87 (UCD-CH31 and UDP96-005). UCD-CH31, UDP96-001 and UDP96-005 were the markers with the highest informative value with regard to the lowest PI (0.06) value, whereas the least informative locus was UCD-CH17 (PI = 0.72) (Table 2). Null allele frequency was the highest in loci UCD-CH17 and UCD-CH21 (0.25) and was the lowest in UDAp-404 ( − 0.06) (Table 2).
Genetic similarity measured within the species ranged between 0.05 and 1.00 within P. avium, 0.45 and 1.00 within P. cerasus, 0.55 and 0.80 within P. angustifolia and 0.05 and 1.00 within P. mahaleb accessions. The average similarity ratios within the species in a descending order were P. cerasus (0.77)>P. angustifolia (0.69)>P. mahaleb (0.55)>P. avium (0.37), respectively.
A tree constructed from the SSR data divided the cultivars into two main clusters according to their taxonomic classification. The first cluster included the P. avium and P. cerasus accessions, the first subcluster consisted of P. avium and the second subcluster included P. cerasus. P. avium seemed to be more differentiated. P. cerasus was divided into five groups, the two biggest groups contained many accessions and these accessions seemed to be identical or very closely related. The second main cluster included the P. mahaleb and P. angustifolia accessions and was also further divided into two subclusters: its first and second subcluster consisted of the P. mahaleb and P. angustifolia accessions, respectively. In contrast to P. cerasus, P. mahaleb and P. angustifolia seemed to be more differentiated. The reference rootstocks were also clustered with their associated botanical species (Fig. 1).

Fig. 1 Dendrogram of 184 Prunus accessions based on UPGMA analysis using the genetic similarity matrix generated by the Nei and Li similarity coefficient after amplification with ten pairs of microsatellite primers.
Discussion
The results obtained in the present study show that microsatellites could be effectively used for fingerprinting purposes in Prunus. In the present study, ten loci in wild Prunus accessions were assayed. The number of alleles per locus ranged from 10 to 20 with an average of 13.3 putative alleles per locus. Previously, Kacar et al. (Reference Kacar, Iezzoni and Cetiner2005) obtained a total of 37 alleles among ten sweet cherry cultivars by nine SSR primers. Clarke and Tobutt (Reference Clarke and Tobutt2003) used 14 sweet cherry cultivars for SSR analysis and determined two to seven alleles per SSR primer. In addition, Vaughan and Russell (Reference Vaughan and Russell2004) used 16 wild cherry accessions for molecular analysis by using ten SSR primers and they detected two to six alleles. In fact, all tested microsatellite primer pairs worked well and produced variable levels of amplifications. The UCD-CH31 locus developed for cherries was the most polymorphic among the ten loci with the highest effective number of alleles (20 alleles) with the lowest PI value (0.06). The CPSCT010, UCD-CH21 and UDAp-401 loci were the less informative with the lowest allele number. Ercisli et al. (Reference Ercisli, Agar, Yildirim, Duralija, Vokurka and Karlidag2011) also found that UDAp-401 and CPSCTO10 were the less informative loci in wild sweet cherries (P. avium). The results showed high amplification of cherry groups with plum, apricot and peach indicating a congeneric relationship within Prunus species. Ercisli et al. (Reference Ercisli, Agar, Yildirim, Duralija, Vokurka and Karlidag2011) successfully used SSR markers identified in other Prunus species to study genetic diversity in wild sweet cherries. Dirlewanger et al. (Reference Dirlewanger, Cosson, Tavaud, Aranzana, Poizat, Zanetto, Arús and Laigret2002), Wunsch and Hormaza (Reference Wunsch and Hormaza2002) and Bouhadida et al. (Reference Bouhadida, Casas, Gonzalo, Arús, Moreno and Gogorcena2009) also showed transportability of SSR markers across Prunus species. Our results demonstrated the cross-species transferability of SSR primers developed in cultivated species to wild species in Prunus for the discrimination of accessions.
Previously, UCD-CH31 (Struss et al., Reference Struss, Ahmad, Southwick and Boritzki2003) and PS12A02 loci were found to be the most informative in other studies (Downey and Iezzoni, Reference Downey and Iezzoni2000; Gulen et al., Reference Gulen, Ipek, Ergin and Akcay2010; Ercisli et al., Reference Ercisli, Agar, Yildirim, Duralija, Vokurka and Karlidag2011). According to Sefc et al. (Reference Sefc, Lopez, Lefort, Botta, Roubelakis-Angelakis, Ibanez, Pejic, Wagner, Glossl and Steinkellner2000), the PI value should be over 0.05 and all loci used in this study had PI values higher than 0.05, indicating that the selected loci were highly polymorphic for used materials. As mentioned before, null allele frequency was the highest in loci UCD-CH17 and UCD-CH21 (0.25) and was the lowest in UDAp-404 ( − 0.06) (Table 2). Null alleles are generally referred to as alleles that fail to amplify during the PCR.
The overall genetic diversity within the tested species was relatively low as evident from the polymorphic ratio of 21% found using SSR primers (Struss et al., Reference Struss, Ahmad, Southwick and Boritzki2003), 19% reported by Zhou et al. (Reference Zhou, Kapel, Hampson, Wiersma and Bakkeren2002) and 20% reported by Zhou et al. (Reference Zhou, Kapel, Hampson, Wiersma and Bakkeren2002) in cherries. We found a high diversity ratio within P. avium compared with other species. A higher level of polymorphism was expected in sweet cherry due to its predominant self-incompatibility (Hegedus et al., Reference Hegedus, Lenart and Halasz2012).
SSR markers have been widely used for molecular characterizations and similarity relationships among Prunus accessions and revealed a high level of polymorphism to discriminate among these accessions (Sosinski et al., Reference Sosinski, Gannavarapu, Hager, Beck, King, Ryder, Rajapakse, Baird, Ballard and Abbott2000; Dirlewanger et al., Reference Dirlewanger, Cosson, Tavaud, Aranzana, Poizat, Zanetto, Arús and Laigret2002; Pedryc et al., Reference Pedryc, Ruthner, Herman, Krska, Hegedus and Halasz2009).
The higher levels of within-group variation observed within P. avium accessions suggest the development of P. avium in the Black Sea and Northeast Anatolia. Introduction and spread of wild and semi-domesticated Prunus species, especially from its native Near Eastern range, domestication of indigenous wild Prunus species, natural hybridization between indigenous and introduced plants, and human selection may have contributed to this high variation.
The H o and H e averaged over the ten SSR loci were, respectively, 0.57 and 0.81, indicating higher mean values than those reported for SSRs in Prunus species (Aranzana et al., Reference Aranzana, Pineda, Cosson, Dirlewanger, Ascasibar, Cipriani, Ryder, Testolin, Abbott, King, Iezzoni and Arús2003; Bouhadida et al., Reference Bouhadida, Casas, Gonzalo, Arús, Moreno and Gogorcena2009). A high allele number and high heterozygosity obtained in the present study reflect the ability of SSR markers to provide a unique genetic profile for individual plant accessions, in particular, for P. avium, P. mahaleb and P. angustifolia accessions.
Such high levels of heterozygosity are commonly observed among clonally propagated, outbreeding, perennial species since they are favoured during selection and are known to confer greater adaptability, vigour and productivity on clonal varieties (Aradhya et al., Reference Aradhya, Liana, Zee and Manshardt1998; Sefc et al., Reference Sefc, Lopez, Lefort, Botta, Roubelakis-Angelakis, Ibanez, Pejic, Wagner, Glossl and Steinkellner2000).
Conclusion
In conclusion, the gene pool of the Prunus species surveyed in Northeast Anatolia has significant amounts of genetic variation. With regard to germplasm management, our results show that the germplasm collection is highly variable and most variation is common to all genetic groups identified. The Prunus germplasm from the region would have economically important adaptive traits that can potentially be incorporated into Prunus breeding programmes. Hence, it is expected that the results of this study will assist current Prunus rootstock breeding efforts in Turkey as well as maintain the genetic integrity of the genetic resources. The SSR-based phylogeny was also generally consistent with Prunus taxonomy based on molecular evidence, suggesting the applicability of SSR analysis for genotyping and phylogenetic studies in the Prunus genus.





