


Females with Turner's syndrome have an increased risk of congenital cardiac defects, hypertension, ischaemic heart disease, and stroke.1 In addition to the familiar obstructive left-sided defects, recent cross-sectional studies have better delineated venous and arterial malformations,2–6 and clinical series describe the advent of sudden aortic dissection.5 When seen in Turner's syndrome, aortic dissection seems to be preceded in most cases by dilation of the aorta, which is influenced by hypertension.7 Dilation of the large arterial conduit vessels may also contribute,8, 9 although a few women may present without risk factors.10 There is little disagreement that females with Turner's syndrome should be monitored clinically not only during childhood and adolescence, but also during adulthood so as to avoid potentially lethal cardiovascular complications. There is, however, no consensus regarding which modalities to use, and the frequency of follow-up visits, in order to detect and prevent aortic dissection. So far echocardiography has been used in most clinics, but recent studies suggest that magnetic resonance imaging of the heart and great vessels might be a superior methodology.2 Further-more, the epidemiology of aortic dissection in Turner's syndrome has not been studied. In the present study we describe 18 cases, mostly occurring early in life, characterized with aspects of genetics, clinical and histological findings of aortic dissection among females with Turner's syndrome.
Material and methods
We identified cases with aortic dissection occurring between 1973 and 2006. Cases were ascertained through Danish national registries,1 through professional contact with patients in Denmark, and through personal contact with all University clinics in Sweden. In Denmark, we searched The Danish Register of Causes of Death, which contains computerized information from certificates relating to all deaths occurring in Denmark since 1973, including date of death, up to 4 causes of death registered using the International Classification of Diseases, version 8 and 10, and manner of death, where there are 5 possibilities, namely natural, accident, suicide, homicide, and unexplained if no information is available.11 The most recent update covers the period 1973 to 1999. Our study was approved by the Danish Data Protection Agency and the involved registers.
All cases of aortic dissection were characterized according to the Stanford classification, where type A dissections involves the ascending aorta regardless of site of the initial tear, and type B include all dissections not involving the ascending aorta.12
All patients had either undergone surgery, or else a post-mortem examination was performed. Some patients had both surgery and an autopsy. Histologic biopsies had been taken in 6 cases, and were re-examined by an experienced cardiac pathologist (UB). Biochemical examination of tissue blocks was performed in 7 cases. Tissue was shaken for 24 hours in a mixture of chloroform and methanol at ratios of 2 to 1 at room temperature in a volume of 3 millilitres. The tissue was dried for 24 hours. The dried tissue was crushed and placed in eppendorf tubes. The dry weight was noted. An equal volume of pepsin was added, along with 1000 microlitres of acetic acid at 0.5 molar concentration. The suspension was left for 21 hours at 4 degree, and thereafter for 3 hours at room temperature. The suspension was centrifuged for 15 minutes at 12,000 revolutions per minute. To the supernatant acetone was added to a final concentration of 50% and left for 18 hours at −20 degree Celsius. 100 microlitres of the sample was added to the ELISA well in a dilution of 1/10, and dried for 24 to 36 hours. Biotinylated goat antibody toward collagen type I and III was supplemented to the dried wells, followed by addition of streptavidin-HRP. The results are expressed as percentages.
Statistical analysis
To estimate an incidence rate of aortic dissection in the 11 Danish women with Turner's syndrome using standard epidemiological methods, we used data from the Danish Central Cytogenetic Registry.13 Here, 783 females with Turner's syndrome were identified by the end of 2001, and their total risk time calculated from birth was 27,495 years. Risk time was assessed from birth, and not from the date of diagnosis of the syndrome. The estimates given are thus the most conservative. We did not include the Swedish patients in these calculations, since we were not able to examine Swedish registers for cases with aortic dissection and Turner's syndrome.
Results
The median age at onset of aortic dissection was 35 years, ranging from 18 to 61 years. All cases are presented in Table 1. Echocardiography, which was performed prior to the acute occurrence of dissection in 10 of 18 cases, showed signs of congenital heart disease with either bifoliate aortic valves, dilation of the aortic root, previous aortic coarctation, or normal conditions. In 5 patients, the findings unequivocally identified bifoliate aortic valves. Hypertension was apparent clinically in 4 of the 18 patients, albeit that no information was available in 2 patients. Of the 18 patients, 10 died from aortic dissection, of type A in 6 patients, type B in 3, while in the final case the site of dissection could not be determined. Our second patient had given birth to twins about one year prior to the aortic dissection. The pregnancy was the result of egg donation. Shortly after being admitted to hospital, she underwent surgery, but it was not possible to fit a graft due to extreme fragility of the remaining aortic tissue, and she subsequently died. Our eighth patient, who was pregnant spontaneously, had the dissection diagnosed during the 7th month. She gave birth by caesarean section, and later a repair was performed, and she is still alive.14 Our ninth patient presented originally with coarctation of the aorta, a hypoplastic arch having been identified in childhood. A repair was performed, but subsequently a pseudoaneurysm developed. She declined re-operation. A year later, she was admitted acutely with signs of dissection. Re-operation was unsuccessful. Our fifteenth patient was diagnosed accidentally during participation in a scientific study, being without symptoms at the time. Approximately one year previously, she had been admitted to hospital with sudden dyspnoea and severe oppression in the upper part of the chest. She was diagnosed with pneumonia and recovered. During the stay in hospital, moderate aortic insufficiency was found. She has now undergone successful surgery for her type A dissection, which extended down to the renal arteries. Our sixteenth patient is the only one thus far who has not undergone surgery. She has been followed for 20 years in a clinic dedicated to Turner's syndrome, with two echocardiograhic examinations having been performed prior to the dissection, the latest in 2001, where the only noted abnormality was slight diastolic dysfunction. Hypertension had not been found prior to dissection. At the age of 61 years the patient was admitted acutely to hospital, and a type B dissection was found starting just distal to a left subclavian artery and stretching below the renal arteries, as well as a bifoliate aortic valve, which had not previously been observed. Our eighth14 and twelfth patients carry the rarer karyotype 45,X/46,XY, but fulfilled the criterions for the clinical diagnosis of Turner's syndrome, and are therefore included in the current study.
Table 1. Clinical characteristics of Turner's syndrome patients with aortic dissection.

The classical karyotype, 45,X, was present in 14 patients, and congenital malformations were present in all but one of those examined pre-morbidly by echocardiography.
Biochemical analysis showed that collagen type I amounted to 61.5%, with a standard error of the mean of 13.5%, while the amount of collagen type III was 38.5%, with a standard error of the mean of 8.9%. Histology showed cystic media necrosis in 3 of 7 cases. All histological data are presented in Table 2.
Table 2. Histological findings in the Turner's syndrome patients.

We calculated a crude estimate of the incidence of aortic dissection in Turner's syndrome. Of the patients collected in this review, 11 are Danish, giving an incidence rate of 40 per 100,000 Turner's syndrome years. Among 0 to 19, 20 to 29, 30 to 39, and 40+ year olds the incidence rates were 14, 73, 78 and 50 per 100,000 years, respectively (Fig. 1). At any age in the entire population in Denmark with Turner's syndrome, aortic dissection occurred in 11 of 783 females, or in 1.4 per 100 females with Turner's syndrome.
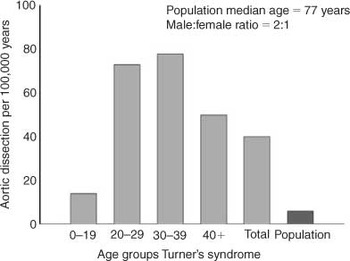
Figure 1. The incidence of aortic dissection per 100,000 years in women with Turner's syndrome and in the general population. Gray bars illustrate females with Turner's syndrome in different age groups, and the dark gray bar indicates the total incidence rate for the entire background population.14,15
Discussion
The most striking finding in our series of patients is the young age at which aortic dissection takes place in setting of Turner's syndrome. The incidence of aortic dissection in the population has been estimated to be 6 per 100,000 per year, with a female median age at occurrence of 77 years,15 and with a ratio of males to females of 2 to 1.16 The average age range for aortic dissection is 50 to 80 years,15 in comparison with a median age of 35 years in our patients. Aortic dissection in that age range is seen only in patients with connective tissue disorders, like Marfan and Ehlers-Danlos syndromes, which are also conditions with increased risk of dilation, aneurysm, and rupture. The most consistent risk factor for aortic dissection is arterial hypertension, which is present in nine-tenths of cases,16 as well as congenital bifoliate or unicommissural aortic valves,17, 18 and coarctation of the aorta.18, 19 In addition, pregnancy, trauma, and iatrogenic-induced trauma are risk factors.18, 19 Amongst our patients, a history of hypertension was present in one-third. In a British study, dilation of the aortic root was seen in two-fifths of 38 women with Turner's syndrome, in association with elevated systolic blood pressure.7 Lately, increased diameters of the great arteries, as well as increased carotid intimal thickness, has also been documented.8, 9 How these findings affect the development of aneurysms and dissection is at present not clear. Importantly, some patients did not have any known risk factors besides Turner's syndrome prior to dissection, possibly due to the fact that they had not had a thorough cardiovascular examination performed.
The distribution of karyotypes, with three quarters of our patients having the 45,X karyotype, differs from the pattern seen in a typical population of females with Turner's syndrome, where approximately half have the 45,X karyotype.13 Of our patients, 2 had the 45,X/46,XY karyotype. Karyotypes with Y chromosomal material typically make up only about 5% of the total population with Turner's syndrome.13 Sybert,5 and Gotzsche et al.,6 also reported a highly increased risk of cardiovascular malformations associated with the 45,X/46,XY karyotype. The karyotype 45,X/46,XY may result in a multitude of clinical phenotypes, and is often classified as “mixed gonadal dysgenesis”, ranging from a normal fertile male via a female with classic features compatible with Turner's syndrome to a normal fertile female. The designation “Turner's syndrome” is a clinical characterization. Today, no firm guidelines exist for the diagnosis,20 but most agree that the cardinal stigmas include retardation of growth, with reduced adult height with or without additional phenotypical features, and except in rare cases, also gonadal insufficiency, and infertility. Since the women fulfilled the criterions for a clinical diagnosis of Turner's syndrome, we chose to include the two women in the study. The 45,X karyotype is most often associated with congenital malformations of the aorta, as well as other organs.1, 5
The Danish patients in our study were found in the Register of Causes of Death and through clinical contact, and form the basis for the epidemiological estimates presented. Previously, we have used the National Register of Patients in the study of Turner's syndrome.1 This registry could not be used in the present context, because it is not sufficiently precise for the study of specific diagnoses like aortic dissection,21 but only for the study of categories of diagnoses like “ischaemic heart disease” or “hypertension”. We could, therefore, have missed a few patients with Turner's syndrome who survived an aortic dissection, and had not necessarily surfaced during clinical contact to the relevant departments. The estimate we present, therefore, will be very conservative, and likely to be an underestimate of the real incidence of aortic dissection. It can be presumed, nevertheless, to be unbiased in other respects, and it will be helpful in the future in unravelling the association between Turner's syndrome and aortic dissection. We found a crude incidence of 40 per 100,000 Turner's syndrome years, greatly exceeding the estimate in the background population, with an even higher incidence rate during 20 to 29 years and 30 to 39 years of 73 and 78 per 100,000 Turner's syndrome years, respectively. In other words, at least 1.4 per 100 females with Turner's syndrome will suffer from aortic dissection. We strongly suspect that aortic dissection is grossly over-represented among women with Turner's syndrome.
Turner's syndrome is usually not included as a risk condition in reviews on aortic dissection.16, 22, 23 Presently, no abnormalities of the aortic wall have been identified in Turner's syndrome. Cystic medial necrosis has been described in some cases, but there are no known biochemical or genetic abnormalities. We found a skewed distribution between collagen type I and III towards more collagen type I, which in normal persons normally is distributed with 30% type I and 70% type III collagen.24 Although the numbers are small, and possibly confounded by the fact that we were not able to include an age-matched group of normal women, we believe that the finding may be informative. A study of aborted fetuses with Turner's syndrome revealed abnormal extracellular matrix of nuchal skin, with increased amounts of glycosaminoglycans relative to the proteoglycan, biglycan.25 The biglycan gene is on the X chromosome and haploinsufficiency of the gene may cause decreased expression. If such an abnormality is also present in the aorta, this may contribute to promoting aortic aneurysms, and eventually, dissection. Support for this suggestion could be the relation between the 45,X karyotype and congenital malformations of the heart in Turner's syndrome.5 Bifoliate aortic valves predominate with the 45,X karyotype,5 as does coarctation of the aorta.26 Coarctation of the aorta occurs more than 350 times as frequent in this syndrome in comparison with the general population.1
Spontaneous pregnancy is a rare event in Turner's syndrome.14, 27, 28 Owing to an increasing number of egg donation programmes, more patients can be expected to become pregnant in the future.29 A recent paper describes a high risk of hypertensive disorders during pregnancy occurring in 5 of 8 completed pregnancies, as well as premature delivery occurring in half.30 Due to the changes associated with pregnancy in blood pressure, cardiac work load, and so on, the risk of aortic dissection is likely to be increased, and two of our patients were either going through a pregnancy, or had done so within the last year. Uneventful cases of pregnancy have been reported,27, 31, 32 albeit that fatal and non-fatal cases due to aortic dissection have also been described.14, 33–36 It seems justifiable to keep pregnant women with Turner's syndrome, and women desiring pregnancy, closely monitored.37
Aortic dissection is a serious complication in Turner's syndrome. Patients with Turner's syndrome should be offered a protocol for clinical follow-up similar to that offered the patients with Marfan syndrome, and each clinic should establish a suitable programme for follow-up.38 With our current knowledge, it seems prudent to keep blood pressure as tightly controlled as possible. Prospective studies are needed in order to establish the exact risk of aortic dissection, which imaging modalities to use,39, 40 to identify patients with an elevated risk, and, if possible, to introduce procedures and/or medicine to lower the risk, both during pregnancy and normal life.
Acknowledgements
The study was supported by a grant from the Danish Health Research Council, grant number 9600822 (Århus University – Novo Nordisk Centre for Research in Growth and Regeneration), and Frimurare-Barnhusdirektionen, Göteborg, Sweden. The expert secretarial assistance of Line Gether is acknowledged.



