Introduction
Phosphoglucose isomerase gene (pgi) encodes the glycolytic enzyme phosphoglucose isomerase (PGI), a dimeric enzyme responsible for the second step of glycolysis and catalyzing the reversible isomerization of glucose-6-phosphate and fructose-6-phosphate. This important metabolism enzyme has been documented to be polymorphic in nearly every studied species, and this polymorphism is maintained by strong natural selection in the wild (Katz & Harrison, Reference Katz and Harrison1997). Earlier studies on biochemical and fitness effects of PGI genotypes showed that genetic variation in this locus was correlated with variation in individual performance and fitness in a wide range of taxa (Pough, Reference Pough1989). Interest in genetic variation at PGI allozyme loci has considerably increased over recent decades. More recent field and laboratory studies in butterflies and beetles have documented strong and consistent genotypic effects on individual performance in terms of flight metabolic rate and dispersal rate in the field (Haag et al., Reference Haag, Saastamoinen, Marden and Hanski2005; Wheat et al., Reference Wheat, Watt and Boutwell2005; Niitepold et al., Reference Niitepold, Smith, Osborne, Reynolds, Carreck, Martin, Marden, Ovaskainen and Hanski2009; Mitikka & Hanski, Reference Mitikka and Hanski2010), temperature adaptation and cold-stress resistance (Dahlhoff & Rank, Reference Dahlhoff and Rank2007; Rank et al., Reference Rank, Bruce, McMillan, Barclay and Dahlhoff2007; Dahlhoff et al., Reference Dahlhoff, Fearnley, Bruce, Gibbs, Stoneking, McMillan, Deiner, Smiley and Rank2008; Saastamoinen & Hanski, Reference Saastamoinen and Hanski2008), egg clutch size and hatching rate, survival, growth rate, pupal mass, fecundity, lifespan and other fitness-related traits (Hanski & Saccheri, Reference Hanski and Saccheri2006; Saastamoinen, Reference Saastamoinen2007; Karl et al., Reference Karl, Schmitt and Fischer2008, 2010; Klemme & Hanski, Reference Klemme and Hanski2009; Saastamoinen et al., Reference Saastamoinen, Ikonen and Hanski2009) and even population growth rate (Hanski & Saccheri, Reference Hanski and Saccheri2006). Therefore, pgi gene can be considered as an important locus underpinning evolutionary adaptation (Karl et al., Reference Karl, Hoffmann and Fischer2010).
The fall webworm, Hyphantria cunea (Drury) (Lepidoptera: Arctiidae), is an important quarantine pest in many parts of the world (Ji et al., Reference Ji, Xie, Li, Gao and Li2003). It was accidentally introduced from North America into Europe in 1946, and now likely occupies Europe from France to the Caspian Sea in the east (Kharazipour et al., Reference Kharazipour, Schöpper, Müller and Euring2009). It was also introduced into Japan in 1945 and then spread into Korea and China (Gomi et al., Reference Gomi, Muraji and Takeda2004; Kharazipour et al., Reference Kharazipour, Schöpper, Müller and Euring2009). Although the fall webworm is usually of only minor economic importance as a forest pest in its native region, it is considered a pest worse than the gypsy moth (Lymantria dispar (Linnaeus)) in invaded regions (Bambara & Baker, Reference Bambara and Baker2006). H. cunea can feed on over 600 species of broadleaf trees and shrubs (Bambara & Baker, Reference Bambara and Baker2006). In China, 175 species of plants in 108 genera of 49 families can be eaten by the caterpillars of H. cunea, including plant species in nearly all taxa of cultivated trees, flowers and crops (Yang & Zhang, Reference Yang and Zhang2007). The caterpillars form webs and feed gregariously on leaves, eventually devouring the entire canopy of their host trees. There are two races of the fall webworm existing in its native region: one with a black head and another with a rusty-orange head in larval stages. Only the black headed race has been introduced into China. Fecundity of this moth is very high, with an average of 800–900 eggs per female and a maximum of 2000 eggs laid in a mass on the underside of a leaf (Sankaran et al., Reference Sankaran, Murphy and Sreenivasan2003). This moth also has a strong potential for dispersal. Males of H. cunea have the potential to fly over 7 km over 12 h on average with a maximum distance of greater than 23 km during this time (Yamanaka et al., Reference Yamanaka, Tatsuki and Shimada2001). The fall webworm has strong temperature tolerance and ecological adaptation. The distribution of this species in its native range is broad, from Canada to Mexico. It was also reported that the pupa of the fall webworm in Japan could survive exposed to −5°C for two weeks, whether it was in a diapause or non-diapause state, and its supercooling point (SCP) reached near −22.9°C (Li et al., Reference Li, Goto, Ito, Sato, Sasaki and Goto2001). It was also found that some life-history traits of the fall webworm had changed following its invasion of Japan, suggesting that it readily adapts to local climates (Gomi, Reference Gomi2007; Gomi et al., Reference Gomi, Adachi, Shimizu, Tanimoto, Kawabata and Takeda2009). Moreover, eradication and control of the fall webworm is arduously difficult. In China, it was firstly introduced into Dandong, Liaoning Province in 1979 and then spread rapidly and broke out in different regions. In 1998, a national control project for the fall webworm was initiated by the SFA (State Forestry Administration) to reduce its population and prevent its further spread and expansion into Beijing (the Capital of China). Although some locations formerly infested by the fall webworm were eliminated and the movement of its infestation was slowed down to a certain extent, new outbreaks are occurring continuously. Based on the data of SFA, 8000 km2 forests were damaged in 116 counties of six provinces (including Liaoning, Hebei, Beijing, Tianjin, Shandong, Shaanxi Provinces) in 2009. At present, the moth is still spreading towards the south, west and north in China. The result of a risk assessment showed that the regions of 21.20°N∼46.33°N, 97.80°E∼132.11°E and 36.81°N∼41.85°N, 76.00°E∼94.66°E were potential distribution areas (Li et al., Reference Li, Gao, Zhang, Ning and Qu2009). This potential range in China is extensive.
As the pgi gene is documented to correlate with insects' fitness and adaptation, in this study, we try to know how many alleles and genotypes of PGI existing in the Chinese population of the fall webworm, in order to further investigation to which genotype has the greatest fitness and potential for ecological adaptation in the future. We first cloned and sequenced full-length pgi cDNA, then detected the diversity and distribution frequency of PGI genotypes and alleles in natural populations based on the electromorphic profile of PGI allozymes and compared the variation of loci among alleles. Also, we displayed the gene structure of PGI in the genome of the fall webworm and compared this to the gene structures of PGI in different species of insects. Moreover, we quantified pgi mRNA expression levels in different stages of the H. cunea's life history. Our goal in this study was to provide basal information for further study on the role of genetic variation of PGI enzymes loci on fitness and adaptation of the fall webworm.
Experimental procedures
Insect material
A colony of H. cunea caterpillars was collected from Beijing in 2008 and reared at 25°C (with a photoperiod of 16L:8D) on an artificial diet provided by the laboratory of Xishan Experimental Forest Farm, Beijing (Zhang et al., Reference Zhang, Wang, Xu and Qu2005). The population has been maintained as an experimental population. All material used in this study were from the experimental population, except the material used for allozyme analysis and allele detection, which came from individuals sampled in the field from five geographic regions (Beijing, Tianjing, Hebei, Shandong and Liaoning Provinces; fig. 1) in 2008–2009. The caterpillars collected from the field were deep frozen and kept at −80°C after emptying their intestines by starving for 24 h.

Fig. 1. Map of geographic distribution of the fall webworm in China, with sampling sites in this study. Dark gray areas represent current distribution regions, and gray areas represent potential distribution regions based on Li et al. Reference Li, Gao, Zhang, Ning and Qu(2009). White dots represent sampling sites. BJ, Beijing; LN, Liaoning; SD, Shandong; HB, Hebei; TJ, Tianjin.
RNA isolation and cDNA cloning of H. cunea's pgi
Total RNA of H. cunea was isolated from each individual during the final larval stage using TRIzol® reagent (Invitrogen Inc., Carlsbad, CA, USA) following the standard protocol. The total RNA was reverse transcribed to the first strand of cDNA using SupertScript™ III First-Strand Synthesis System kit (Invitrogen). Based on the analysis of pgi mRNA sequences in Colias eurytheme (Boisduval) and Bombyx mori (Linnacus), a pair of degenerate primers (PGI-L, PGI-R) were designed for amplifying a partial cDNA sequence of H. cunea's pgi by RT-PCR (reverse transcription polymerase chain reaction). Then, the 3′-end sequence was obtained by RACE (rapid amplification of cDNA ends) using a 3′-Full RACE Core Set Ver. 2.0 kit (TaKaRa Bio Inc., Shiga, Japan) with primers PGI-3-OUT and PGI-3-IN, and the 5′-end sequence was also obtained by RACE using a SMART™ RACE cDNA amplification kit (Clontech Laboratories Inc., Terra Bella, CA, USA) with primer PGI-5, both following the manufacturer's protocols. All PCR products were cloned into PGM-T vector (Tiangen Biotech Co. Ltd, Beijing, China) after a gel extraction with a EZ Spin Column DNA Gel Extraction kit (Sangon Biotech Co. Ltd, Shanghai, China) and then sequenced by Invitrogen (Shanghai Invitrogen Biotechnology Co. Ltd, Beijing, China). In addition, two pairs of primers (full-pgi-L-OUT/full-pgi-R-OUT and full-pgi-L-IN/full-pgi-R-IN) were designed and used to amplify the full length of the coding region of H. cunea's pgi. All primers used for cDNA amplification are listed in table 1.
Table 1. List of primers used in this study.

Isolation of the genomic DNA and amplification of H. cunea's pgi
Genomic DNA of H. cunea was extracted from the final larval stage using a phenol chloroform extracting method. Referring to the orthologous pgi genomic sequences of C. eurytheme and B. mori, 11 pairs of primers were designed for pgi intron amplification from H. cunea's genomic DNA. PCR was carried with 94°C 3 min, then 35 cycles of 94°C 30 s, annealing with an appropriate temperature, 30 s and 72°C 2 min, and finally extension at 72°C 10 min. All PCR products were cloned into PGM-T vector after a gel extraction and then forward and reverse sequenced by Invitrogen Inc. Some inner primers were designed for sequencing lengthy introns. The fragment sequences were assembled using the software Sequencher™ 4.1.4 to get the full length of pgi genomic sequence of H. cunea. To verify whether the assembled sequence was corrected or not, verification PCR was performed with different primer-pairs using LA Taq polymerase (TaKaRa Bio Inc., Shiga, Japan) and with an extension time of 15 min cycle−1 and a 25-min final extension. The validity of PCR products was judged based on the fragment sizes on electrophoretic agarose gel.
Quantitative real-time RT-PCR
A quantitative real-time RT-PCR (RT-qPCR) was performed to compare pgi mRNA expression levels in each stage of H. cunea's life history. Total RNA was extracted from each stage of the fall webworm (eggs, 1st to 6th instar larvae, pupae and adults) with Trizol reagent and used for the first strand synthesis. A pair of primers (qPCR-pgi-L/qPCR-pgi-R) was used for the qPCR with 25 μl of mixture (containing cDNA template, primers and SYBR® Premix Ex Taq™ (TaKaRa) and ROX™ Reference Dye II (TaKaRa)) on an ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) by the following cycling parameters: 10 min at 95°C; 40 cycles of 15 s at 95°C and 1 min at 60°C. Actin gene of H. cunea with primers (qPCR-actin-L/qPCR-actin-R) was used as internal control to normalize gene expression level. Melting curve analyses were performed immediately using the instrument's default setting after each PCR. The mean Ct value was obtained from four replicas of each cDNA template in each RT-qPCR. The experiments were repeated three to five times from RNA preparation to RT-qPCR, and each cDNA template was performed with RT-qPCR five times. The mean value of all repeats was used to analyzing the gene expression level of each stage. SPSS software was used to analyze the statistical difference among expression levels in different stages.
Allozyme analysis
For polymorphic analysis, each individual was divided into two parts: one for allozyme analysis and the other for RT-PCR. Each half-individual was homogenized in Tris-HCl grinding buffer-PVP solution (buffer contains 0.1% v/v 2-mercaptoethanol, 0.001 M EDTA (tetrasodium salt), 0.010 M potassium chloride, 0.010 M magnesium chloride hexahydrate, 14% w/v PVP 40 000, 0.10 M Tris-HCl buffer, pH 7.5.) (Soltis et al., Reference Soltis, Haufler, Darrow and Gastony1983). Vertical slab polyacrylamide gel (3% spacer gel and 7% separation gel) electrophoresis was performed using a glycin electrophoretic system. PGI enzyme staining protocol followed Soltis (Reference Soltis1989). The polyacrylamid gel was stained 20 min with a staining buffer (50 ml of staining buffer containing 50 mM Tris-HCl PH 8.0 50 ml, NAD 10 mg, fructose-6-phosphate Na2-salt 20 mg, glucose-6-phosphate dehydrogenase 20 units, MTT 10 mg and PMS 2 mg). The genotypes were inferred based on the segregation pattern characteristics of a dimeric codominant enzyme: those with a single-band phenotype were suggested as one locus in homozygotes in control of a dimeric enzyme with two homodimers, and those with a triplet phenotype were suggested as one locus in heterozygotes in control of a dimeric enzyme with a centrally migrating heterodimer. The pgi alleles were named based on electrophoretic mobility, with pgi-a for one with minimum mobility, pgi-b for one with secondary mobility, and so on. Individuals with homozygotes were also used for allele determination by RT-PCR and sequencing.
Sequence analysis
To make sure the correction of each sequence, forward and reverse sequencing were performed for each fragment. Sequences were assembled using Sequencher™ 4.1.4. Orthologous sequences of other insects’ pgi were downloaded from NCBI (http://www.ncbi.nlm.nih.gov/). The ClustalW program was used for sequence alignment. DnaSP5.10.01 (http://www.ub.es/dnasp) was used for sequence polymorphism analysis. Phylogenetic analysis was performed by Mega 4 software using bootstrap method (resampling 1000). Prosite (http://expasy.org/cgi-bin/prosite) was used for protein domain analysis. ProtParam (http://expasy.org/cgi-bin/protparam) was used for MW (molecular weight) and PI (isoelectric point) analysis. ELM (http://elm.eu.org/) was used for motif search. Gene structure was drawn using DNAMAN software. All sequences used in this study were listed in table 2.
Table 2. List of species and NCBI sequence ID of its pgi gene used in this study.

* Genomic DNA sequence has not submitted to NCBI; we downloaded it from Silkworm Genome Database (http://silkworm.genomics.org.cn/).
Results
Isolation and characterization of H. cunea's pgi
A total of 2110 bp (base-pair) pgi cDNA full-length was obtained from H. cunea using the RT-PCR and RACE methods, containing a 1671-bp coding region, a 125-bp 5′-untranslated region (UTR) and a 314-bp 3′-UTR. The sequence of 3′-UTR was AT-rich (A+T content 73%), with a putative polyadenylation signal sequence (ATTAAA) at 171 bp upstream of the poly (A) tail (12 bp). The initiating codon (ATG) was at the positions 126–128 of the 5′-end. A sequence (ACTATGG) was formed with the base (A) at the third position upstream from the initiation codon (−3) and G at the position after the initiation codon (+4), conforming to the Kozak rule (A/GXXATGG) (Kozak, Reference Kozak1997).
A 556-aa (amino acids) polypeptide was deduced from the cDNA of H. cunea's pgi, with a theoretical MW 61.5 KDa and pI 6.1. Two phosphoglucose isomerase signature sequences were contained in the deduced protein sequence, i.e. the sequence (DWVGGSYSLWSAIG) at the sites 271–285 and the sequence (GVIWDMNSFDQWGVELGK) at the sites 505–522, and they are putative conserved SIS (sugar isomerase) domains. The deduced protein sequence has high identity with those of other insects’ PGI by BlastP in NCBI, such as 92% identity with Spodoptera exigua, 88% with Melitaea cinxia, 87% with Biston betularia, Colias eurytheme and Euphydryas aurinia, 86% with Bombyx mori and Colias meadii, and 75% with Drosophila pseudoobscura. The cDNA full-length sequence of H. cunea's pgi was submitted to GenBank, with accession numbers: JN191711–JN191746.
The gene structure of H. cunea's pgi in the genome was obtained by PCR amplification from the genomic DNA of the moth. A total of 14,332 bp sequence was obtained, including 12 exons and 11 introns (fig. 2), which was similar to the structure of Bombyx mori's and Colias eurytheme's (fig. 3). The length of exons was from 95 to 188 bp, with A+T content from 43.36% to 64.89%. The length of introns was from 342 to 2000 bp, with A+T content from 58.26% to 75.57%. All introns began with GT and ended with AG. The GT-AG splice sites are spliceosomal introns’ characteristics.

Fig. 2. Scaled diagram of the gene structure of H. cunea's pgi, with the start and stop positions of exons in cDNA and genomic DNA.

Fig. 3. Gene structure of phosphoglucose isomerase in insect's genome. Block represents exon and line represents intron. Arrows indicate homologous regions. Number at the left side is the gene length in genomic DNA of the right species. *, a 5948-bp length that is omitted in Aedes aegypti.
Phylogenetic analysis and structure comparison of insects' pgi
We downloaded all PGI protein sequences of insects deposited in NCBI, which belong to 34 species in five orders. Most of these species belong to Endopterygota which undergo complete metamorphosis. Only two species (pea aphid Acyrthosiphon pisum (Harris) and head louse Pediculus humanus corporis De Geer) belong to Paraneoptera which undergo incomplete metamorphosis. Taking one of the two species as an outgroup, the phylogenetic tree showed that all species with complete metamorphosis were gathered together to form a monophyletic group, and it was divided into five cludes with high bootstrap supports (fig. 4), i.e. Lepidoptera (including four moths and four butterflies), Hymenoptera (including one bee, three ants and one wasp), Diptera (Brachycera) (12 species in Drosophila and one in Glossina), Diptera (Nematocera) (three mosquitoes) and Coleoptera (Tribolium castaneum). Based on the phylogenetic tree, it is inferred that Hymenoptera and Lepidoptera were monophyletic groups. However, Diptera didn't form a monophyletic group. Cyclorrhapha (comprised of flies) and Nematocera (comprised of mosquitoes) each formed an independent monophyletic group. We suppose that it might be due to an adaptation of different glycogen substrates in food leading to the divergence of pgi in the two groups, because mosquitoes feed on animal blood, while flies do not. The result implies that the pgi gene may not be a suitable marker for inferring the phylogenetic relationship of Insecta Orders.

Fig. 4. Phylogenetic tree inferred from amino acid sequences of insects' phosphoglucose isomerase by neighbour joining method. Bootstrap values (1000 replicates) larger than 50% are shown below branches.
The comparison of pgi gene structure among H. cunea and 16 other species showed that these gene structures were obviously different (fig. 3). Although the length of cDNA sequences (from 1668 to 1686 bp) and amino acid sequences (from 555 to 561 aa) of PGI are very similar in different insects, the pgi lengths in genome are dramatically different, from 2395 bp in Drosophila melanogaster to 21,233 bp in Aedes aegypti. Variations mainly occur in introns and splice sites, owing to these regions being less subjected to selection, leading to the number of exons varied among insect groups being from four to 12 (fig. 3). Within each group, however, their structures are very similar and conserved. In mosquitoes, there are four exons with three at 5′-end and one at 3′-end, with a huge intron in the middle. In flies, the gene is small with five exons and four small introns. In Hemiptera (Sternorrhyncha) (aphid) there are ten exons with nine introns, and some introns are large. In Anoplura (lice), there are also ten exons with nine small introns. In Hymenoptera, this gene is composed of 11 exons and ten small introns. In Lepidoptera, 12 exons with 11 large introns are included in this gene. If the structure among the hemimetabolous insects (aphid and lice with ten exons) is taken as a plesiomorphy, the pgi gene structure in insects may have evolved towards either increasing or reducing the number of introns and exons.
Polymorphism of genotypes and alleles of H. cunea's PGI in the Chinese population
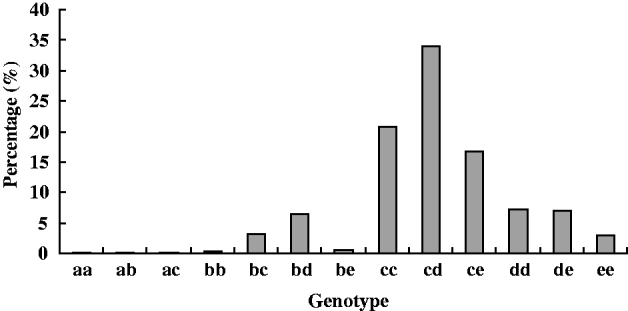
Allozyme analysis was performed on a total of 1891 individuals of H. cunea. Thirteen electrophoretically distinct (electromorph) PGI genotypes were detected, which comprised of five electrophoretic mobility classes (a, b, c, d and e). The distribution frequencies of these genotypes in the natural population were different (fig. 5). Among them, pgi-cd was the most common genotype in the Chinese population, with more than 34% of all genotypes. This membership abundance was followed by pgi-cc and pgi-ce. More than 70% individuals were allocated to these three genotypes. Frequency of genotype pgi-dd, pgi-de, pgi-bc, pgi-bd and pgi-ee was 3–8%. However, genotype pgi-aa, pgi-ab, pgi-ac, pgi-bb and pgi-be were rare with a frequency of each less than 1%.
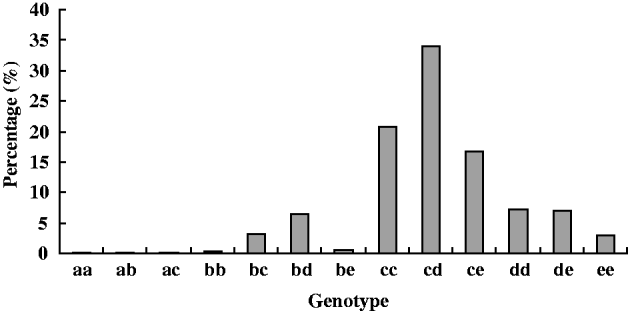
Fig. 5. Genotype frequency of H. cunea's PGI in the Chinese population.
Based on the frequency of genotypes, the distribution frequency of each allele in the Chinese population was computed. Among them, pgi-c was the most prevalent allele (47.83%), followed by pgi-d (31.04%), pgi-e (15.12%) and pgi-b (5.68%). The pgi-a was a rare allele (0.32%) in the Chinese population.
We sequenced the cDNA sequence of each individual having homozygotic pgi genotype and obtained 36 cDNA full-length sequences belonging to four alleles except pgi-a, the later was too rare and we could not get the sequence. Comparing these allele sequences, no deletion and insertion were detected. Thirty stably variable sites were found among the four alleles with 27 sites at the third codon position, one at the second and two at the first. The variable sites are listed in table 3. Most of them are synonymous mutations, and only five sites were non-synonymous mutations that caused amino acid change. Examples of such changes are: nucleotide G/A conversion at site 248 of the cDNA sequence led to genetic code AGG/AAG conversion and amino acid Arginine (R)/Lysine (K) conversion at site 83 of the protein sequence; nucleotide C/G at site 403 of cDNA sequence led to code CAG/GAG and Glutamine (Q)/Glutamic acid (E) at site 135 of the protein sequence; nucleotide A/T at site 732 of cDNA sequence led to code AAA/AAT and Lysine (K)/Asparagine (N) at site 244 of protein sequences; nucleotide C/A at site 882 of cDNA sequence led to code GAC/GAA and Aspartic acid (D)/Glutamic acid (E) at site 294 of protein sequences; and nucleotide A/C at site 1360 of cDNA sequence led to code AAG/CAG and Lysine (K)/Glutamine (Q) at site 454 of protein sequence. These changes were not on motif except the last one (at site 454 in protein sequence), which was at a motif of glycosaminoglycan attachment site (454–457 QSGM/KSGM). As K is hydrophobic and Q is hydrophilic, it is unknown whether the change of hydrophobicity could impact the motif's activity or not.
Table 3. Segregating nucleotide variation among the main alleles of H. cunea's pgi.

* is non-synonymous mutation site.
Beyond these stably variable sites, many accidental point mutations were also observed in the 36 cDNA sequences of H. cunea's pgi. These polymorphic sites composed different haplotypes within each allele. A total of 220 variable sites were detected among these sequences. The total ratio of polymorphic sites was 13.17%. However, most of them (72.27%) were mutations by chance, which only occurred once among all sequences. Sixty-one sites (27.73%) occurred at least twice in all sequences (including the 30 stable variation sites depicted above). The average nucleotide diversity of these sequences was πtotal=0.01560, Theta θ=0.03247, Tajima's D=−1.96087. A negative Tajima's D signifies an excess of low frequency polymorphisms, indicating population size expansion and/or purifying selection (Biswas & Akey, Reference Biswas and Akey2006).
Analysis of the protein sequences deduced from the 36 cDNA sequences revealed that a total of 129 loci were polymorphic. The ratio of polymorphic loci was 23.20% of the total. The average values of the rates of synonymous mutation (Ks) and non-synonymous mutation (Ka) between each two sequences among them were Ks=0.03786, Ka=0.00908, Ka/Ks=0.39496. Ka/Ks value is an important measure of evolutionary direction. A Ka/Ks value less than 1 means that negative selection acts at this locus. Our result indicates that the gene pgi is subject to functional constraint such that non-synonymous amino acid substitutions are deleterious and purged from the population.
Differentiation of H. cunea's PGI in geographical subpopulations
Comparing the distribution frequencies of genotypes and alleles of H. cunea's PGI in five Chinese geographical subpopulations, it was shown that high polymorphism was detected in the subpopulations of Beijing, Liaoning and Shandong, with 9–11 genotypes distributed in each of them. However, in the subpopulations of Hebei and Tianjin, low polymorphism was detected, with only 3–5 genotypes distributed in each of them, respectively. The most predominant genotype in each subpopulation was also different. Pgi-cd was predominant in the subpopulations of Beijing, Liaoning and Tianjin, and pgi-bd in the subpopulation of Shandong and pgi-dd in the subpopulation of Hebei (fig. 6). Also, the frequency of alleles in each geographic subpopulation is different (fig. 7).

Fig. 6. Genotypes frequency of H. cunea's PGI in five Chinese geographic subpopulations. BJ, Beijing; LN, Liaoning; SD, Shandong; HB, Hebei; TJ, Tianjin.

Fig. 7. Allele frequency of H. cunea's PGI in five Chinese geographic subpopulations (▪, a; ![]() , b;
, b; ![]() , c;
, c; ![]() , d;
, d; ![]() , e).
, e).
Pgi mRNA expression profile in H. cunea's life history
Comparison of Comparison of pgi mRNA expression level in each stage (including eggs, 1st to 6th instar larvae, pupae and adults) of H. cunea's life history showed that the highest expression level was in the 6th instar larvae stage, followed by that in egg stage and in adult stage (fig. 8). Expression levels in the 2nd to 5th instar larvae stages and in pupae stage were similar and stable. But the expression level in the first instar larvae stage was lower than those in others. The differences among stages of H. cunea's life cycle were statistically significant (fig. 8). We speculate that the pgi mRNA expression level perhaps reflects the energy demands in the insect's body. Energy demands are distinct in different stages of H. cunea's life cycle. In the egg stage, energy is needed for embryo development. In the last instar larvae stage, energy accumulation is needed for pupation. In the adult, energy is needed for flight, mating and reproduction. Therefore, a large amount of energy is required for their activities, and the energy metabolism is especially vigorous in these stages. This may explain why pgi mRNA expressions levels in these stages were higher than those in other stages. But, in the first instar larvae stage, perhaps because the body is very small and less energy is needed for their activities in this stage, the pgi expression level is lower than that in other stages.

Fig. 8. Pgi mRNA expression levels in different stages of H. cunea's life history. Letter above each pillar represents a statistically significant difference (P<0.05). Bars are standard errors.
Discussion
Alleles and genotypes of PGI are rich and polymorphic in natural population of species. In our study, five alleles were detected in the Chinese population of the moth H. cunea, and the total frequencies of the two prevailing alleles (pgi-c and pgi-d) reached 78.8%. That's similar to the results studied in butterflies. It was reported that populations usually harbor four to six allelic mobility classes in Colias butterflies (Watt, Reference Watt1977). Watt and his associates observed six alleles in Colias and found that the total frequencies of two prevailing alleles reached nearly 90% (Watt et al., Reference Watt, Wheat, Meyer and Martin2003). The same case was observed in Melitaea cinxia. Two alleles pgi-d and pgi-f were common in M. cinxia, and the frequencies of these two alleles varied significantly with population age and connectivity (Haag et al., Reference Haag, Saastamoinen, Marden and Hanski2005). It was suggested that strong positive selection maintains the extensive PGI allozyme polymorphism (Wheat et al., Reference Wheat, Watt, Pollock and Schulte2006).
Thirteen electromorph genotypes comprised of the five PGI alleles were detected in the Chinese population of the moth H. cunea. The distribution frequencies of those genotypes in natural population were very different (fig. 5). The most common genotype was heterozygote pgi-cd (34%), then pgi-cc (20.8%) and pgi-ce (16.5%). More than 70% individuals were allocated to these three genotypes. However, allele pgi-a was very rare, with a total frequencies of three genotypes (pgi-aa, pgi-ab and pgi-ac) detected in the nature population less than 0.5%. Genotype pgi-bb and pgi-be were also rare, with a frequency of each less than 1% in the natural population. Results of early studies have shown that PGI genotypes differ in kinetic parameters and in their fitness (performance) in field and laboratory studies (Carvalho, Reference Carvalho1988; Patarnello et al., Reference Patarnello, Bisol and Battaglia1989; Johannesson et al., Reference Johannesson, Kautsky and Tedengren1990). It was reported that dispersal rate, fecundity and survival were affected by two alleles, A and C, in the butterfly M. cinxia. Individuals with the C allele being more mobile, the AC heterozygotes and the CC homozygotes had higher mobility than the AA homozygotes (Zheng et al., Reference Zheng, Ovaskainen and Hanski2009). Fitness differences were also reported to associate with single-nucleotide polymorphisms (SNPs) of M. cinxia's PGI (Orsini et al., Reference Orsini, Corander, Alasentie and Hanski2008). In the butterfly Lycaena tityrus, it was reported that PGI genotypes affected growth rate and pupal mass significantly (Karl et al., Reference Karl, Schmitt and Fischer2008). In the leaf beetle Chrysomelia aeneicollis, it was observed that directional changes in PGI allele frequency variations were related to HSP70 expression, temperature stress, resistance, running speed, survival and fecundity (Dahlhoff & Rank, Reference Dahlhoff and Rank2000, Reference Dahlhoff and Rank2007). Although we have not yet assessed the fitness of each genotype in the moth H. cunea, we suppose that the role of natural selection acting on those genotypes might be different. It might be positive to the rich genotypes and negative to the rare genotypes. The fitness of different genotypes in the fall webworm is worth further study in the future.
In our result, it was shown that the diversities and distribution frequencies of alleles and genotypes of H. cunea's PGI in the five Chinese geographical subpopulations were different. H. cunea is an invasive pest. High frequency of some alleles is likely to be related with the ‘founder effect’ and genetic drift. Based on our current data, however, it's difficult to determine, because muti-factors could impact allele frequency, such as demographic sampling bias, pesticide utilization, host adaptation, and so on. Moreover, we also tried to find some association between the climatic conditions and the frequencies of alleles in the five sampling geographic regions, as it was reported that frequency of pgi alleles could be related with temperature resistance (Dahlhoff & Rank, Reference Dahlhoff and Rank2000, Reference Dahlhoff and Rank2007). We obtained the climatological data, compared the parameters (including maximum and minimum temperature in a year, total effective temperature in a year and average temperature in the sampling month) and analyzed the correlations between temperature and alleles frequency in different geographic regions. However, no obvious correlation was found between climatic condition and allele frequency (R2<0.3, P>>0.05). Therefore, a further investigation is needed in the future.
Acknowledgements
This work was supported by the National Basic Research Program of China (grant no. 2009CB119200) and Beijing Municipal Natural Science Foundation of Beijing (grant no. 6082013).