Congestive heart failure can be characterized by the sequential and progressive activation of numerous cascades of endocrinal activity. The upregulation of the renin–angiotensin–aldosterone system, as well as increased circulating levels of norepinephrine and endothelin-1, have been well described as playing a significant role in the progression of left ventricular dysfunction, and development of heart failure, in adults.1 An impressive amount of data has therefore accrued rapidly in adults regarding cardiac hormones, but to a lesser extent in children. Neurohormonal activation is characteristic of heart failure. Unlike the aforementioned vasoconstrictor neurohormones, the cardiac hormones atrial natriuretic peptide and B-type natriuretic peptide have beneficial actions, such as vasodilation and natriuresis (Fig. 1).2 Although there has also been an escalation of investigative studies on these cardiac hormones in adults, there is virtually no information of the actions in children. In addition, the significance of the cytokines, and other markers of inflammation, is also being much more recognized in the pathophysiologic state of heart failure. Our aim in the limited review is to provide an account of the fundamentals of the neurohormonal axis, as well as to familiarize the reader with newer concepts in the use of biochemical markers in the progression, diagnosis, treatment, and prognosis of heart failure.
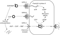
Figure 1.
The interplay between the renin–angiotensin system and the natriuretic peptides. The angiotensin system leads to vasoconstriction, fibrosis, decreased diuresis, and sodium retention while the natriuretic system serves as a counter-regulatory system with natriuresis, diuresis, vasodilatation, antifibrosis, and inhibition of renin and aldosterone. Ang I: angiotensin-I; ANP: atrial natriuretic peptide; AT1R: angiotensin type-1 receptor; BK: bradykinin; BNP: B-type natriuretic peptide; cGMP: cyclic guanosine monophosphate; CNP: C-type natriuretic peptide; GC: guanylate cyclase; NEP: neutral endopeptidases; PDE: phosphodiesterase; RA: particulate guanylate cyclase A receptor; RB: particulate guanylate cyclase B receptor; RC: particulate guanylate cyclase C receptor; K+: potassium ion; NO: nitric oxide; PK: protein kinase. (Reproduced with permission from Burnett JC, et al. Alterations in the kidney in heart failure: the cardiorenal axis in the regulation of sodium homeostasis. In: Mann DL (ed.). Heart Failure: A Companion to Braunwald's Heart Disease. Saunders, 2004.)
Pathophysiology of heart failure
The onset of heart failure, with arterial underfilling manifested by decreased activation of mechanoreceptors in the left ventricle, carotid sinus, aortic arch, and renal afferent arterioles, leads to a myriad of pathophysiologic adaptations, including stimulation of the adrenergic system, activation of the renin–angiotensin–aldosterone system, and release of substances (such as endothelin and vasopressin). As heart failure progresses, however, the excessive expression of these systems leads to severe vasoconstriction, and increases in systemic vascular resistance, retention of sodium and fluid, and eventually myocardial fibrosis and apoptosis. The use of biochemical markers in the overall management of heart failure has become progressively more evident, both in research and in clinical medicine. For markers of heart failure to be useful, however, it is necessary to take note of a few important concepts. A biomarker should be very sensitive and specific. Its use should be economically feasible, the technique should be readily and easily performed, so as to make the resulting information widely available. The inherent error in the technical measurement performed, known as the coefficient of variation, should be low enough that minor changes in the biomarker are representative of clinical changes over the entire spectrum of the severity of disease. A biochemical marker should also make some sense in relation to the clinical state of the patient. The changes evident in the marker should be consistent with the clinical course, and offer insight into the prognosis and possible future issues with regard to management.
Sympathetic nervous system
The ventricular dysfunction that occurs with heart failure results in an increase in sympathetic tone mediated by baroreceptors that has several consequences, including tachycardia, increased myocardial contractility, arterial vasoconstriction with resultant increased afterload, and venoconstriction resulting in increased preload. The increase in sympathetic tone is manifested by increased levels of epinephrine and norepinephrine in the plasma, which mediate the aforementioned biologic effects.
Increased levels of local and circulating norepinephrine may contribute to myocytic hypertrophy, either by direct stimulation of alpha- and beta-adrenergic receptors, or indirectly by activating the renin–angiotensin–aldosterone system. Anand et al.3 in the Valsartan Heart Failure Trial, evaluated patients with moderate to severe heart failure who were treated with Valsartan, a blocker of the receptors to angiotensin-II. Not only was norepinephrine, as well as brain natriuretic peptide, shown to be a predictor of morbidity and mortality, but their decrease over a 2-year period concomitant with treatment with the drug was associated with a corresponding decrease in morbidity and mortality.
The renin–angiotensin–aldosterone system
The renin–angiotensin system is well known for its regulation of blood pressure and homeostasis. It has been known for some time that the system is activated in the setting of heart failure. In patients with progressive heart failure, there are marked increases in circulating levels of renin, angiotensin-II, and aldosterone.4 The excessive adrenergic stimulation, and activation of the renin–angiotensin–aldosterone system, may play a major role in the progression from cardiac hypertrophy to cardiac failure.
Angiotensin-II regulates blood pressure by modulating renal absorption of sodium and water, by stimulating release of aldosterone, or by stimulating the recognition of thirst in the central nervous system. Angiotensin-II is produced by the enzymatic cleavage of angiotensinogen by renin, an aspartyl protease, to form angiotensin-I. The actions of angiotensin-II result from binding to receptors, which are in the family of G-protein-coupled receptors.5 Angiotensin-I mediates vasoconstrictor effects, while angiotensin-II mediates vasodilatory effects. Both of these receptors are present in the kidney, brain, and adrenals. The renin–angiotensin system exists in tissues that can produce and respond to locally generated angiotensin-II. This system plays a critical role in both vascular and cardiac remodeling.6 The renal excretion of sodium and water usually coincides with intake of sodium and water. In patients with heart failure, this is not the case, as there is paradoxical retention of sodium and water, even though the intravascular volume is elevated.
Angiotensin-II may be directly toxic to cardiac myocytes.7 The degree of its activation in the setting of heart failure has been shown to be proportional to the severity of disease, increasing with the overall progression of heart failure, and apparently related to prognosis.8 The clinical implications were emphasized by Benedict et al.,9 who conducted trials showing that treatment with drugs such as angiotensin-converting enzyme inhibitors that decrease overall morbidity and mortality in heart failure also cause an attenuation or reduction in angiotensin-II, as well as other neurohormones.
Aldosterone can be synthesized in vascular smooth muscle and endothelial cells, the brain, blood vessels, and the myocardium. Receptors for aldosterone have been found in the kidney, colon, salivary glands, brain, heart, and blood vessels.10 Aldosterone may cause direct profibrotic effects, and its interaction with angiotensin-II also promotes fibrosis by increasing the expression of plasminogen activator inhibitor-1.11 In humans, elevated levels of aldosterone are associated with endothelial dysfunction, myocardial infarction, left ventricular hypertrophy, and death.12
Natriuretic peptide hormones
It was de Bold,13 using rats, who made the observation that infusion of extracts of atrial tissue results in a copious natriuresis, confirming the endocrinologic properties of the heart. This ultimately led to the isolation and reproduction of atrial natriuretic peptide. This peptide was the first member of a family of peptides subsequently shown to have natriuretic, diuretic, and vasorelaxant properties.14 These natriuretic peptides are a group of structurally similar proteins that are genotypically distinct. They constitute a family of vasoactive agents, with many favorable physiologic actions, which have become the subject of many diagnostic tests and therapeutic interventions in adult cardiology. Three major natriuretic peptides, all of which share a 17-amino-acid ring, are commonly accepted: namely the atrial natriuretic peptide, B-type natriuretic peptide, and C-type natriuretic peptide. The precursor hormone for each peptide is encoded by a separate gene, and the regulation of each protein is unique. These peptides act on a multitude of organs via guanylate cyclase-linked membrane receptors that produce natriuresis, diuresis, and vasodilation.
Natriuretic peptides interact with high-affinity receptors on the surface of target cells. Three different receptors, one each for the peptides, have been identified in mammalian tissue (Fig. 2). Receptors A and B are linked to cascades mediated by the cyclic guanosine monophosphate signaling system, which is related to many of the renal and cardiovascular effects of the natriuretic peptides. The A and B receptors are similar in structure, with approximately two-fifths homology in the ligand-binding extracellular domain.15 The A-type receptor binds both atrial natriuretic peptide and brain natriuretic peptide, with a higher affinity for the atrial peptide. The B-type receptor binds C-type natriuretic peptide. The B-type receptors predominate in the brain, while the A-type receptors are more abundant in large blood vessels. Both A and B types are present in the kidney and adrenal glands. The extracellular portion of the receptor is linked to the intracellular portion by a single membrane-spanning section. This intracellular portion contains a guanylyl cyclase catalytic domain that leads to the elevation of cyclic guanosine monophosphate. The C-type receptor is responsible for the clearance of natriuretic peptides. It is a homodimer protein, of which each monomer has a membrane-spanning segment. The natriuretic peptides, once bound, are internalized and degraded, with return of the C-type receptor to the cell surface. Natriuretic peptides can also be degraded by cleavage of neutral endopeptidases that are present in renal tubular cells and vascular cells.
Figure 2.
Natriuretic peptides and receptors. The natriuretic peptides, ANP, BNP, and CNP, have different interactions with the receptors for natriuretic peptides. While ANP and BNP interact mainly with NPR-A, CNP relates more to NPR-B. Note that NPR-C (as well as neutral endopeptidases) is responsible for the degradation of all natriuretic peptides. ANP: atrial natriuretic peptide; BNP: B-type natriuretic peptide; CNP: C-type natriuretic peptide; NPR-A, B, and C: natriuretic peptide receptors A, B, and C; GTP: guanosine triphosphate; cGMP: cyclic guanosine monophosphate. (Reproduced with permission from de Lemos JA, et al. B-type natriuretic peptide in cardiovascular disease. The Lancet 2003; 362: 316–322.)
Atrial natriuretic peptide
Atrial natriuretic peptide is of cardiac origin. It is a cyclic 28-amino-acid polypeptide synthesized predominantly from the atrium of the normal heart. It is stored in atrial granules as a 126-amino-acid prohormone known as pro-atrial natriuretic peptide.16 Following secretion, serine protease corin splits pro-atrial natriuretic peptide1–126 in equimolar amounts into an N-terminal fragment, consisting of 98 amino acids, and biologically active atrial natriuretic peptide.17 Both fragments circulate in the plasma. There is suggestion that the N-terminal fragment has biologic actions similar to those of the peptide itself.18 The peptide is rapidly removed from the circulation via hydrolysis and activity of specific receptors.19
Levels of atrial natriuretic peptide, and its prohormone, are elevated in the setting of heart failure and volume overload because increased tension, and resultant stretch on the atrial walls, leads to activation of the cardiac hormonal system.20 Levels of the peptide are relatively closely related to left atrial pressures, and can be released into the bloodstream following a minor stimulus such as exercise.21 There appears to be a possible link between elevated levels of atrial natriuretic peptide and moderate to severe rejection following orthotopic heart transplantation.22 Levels of atrial natriuretic peptide, and B-type natriuretic peptide, have been shown to be augmented in the setting of myocarditis.23 In animal studies, when given in low doses, infusions of the peptide lower blood pressure, and reduce peripheral vascular resistance. Infusions at higher doses result in increases in peripheral vascular resistance, suggesting counter-regulatory activation of baroreceptors.24 Human studies are currently underway.
In the kidney, atrial natriuretic peptide, as well as B-type natriuretic peptide, increases glomerular filtration, and inhibits the reabsorption of sodium, causing a natriuresis and diuresis.25 Atrial natriuretic peptide also causes the relaxation of vascular smooth muscle, which ultimately leads to arterial and venous dilation, with resultant reduction in ventricular preload and blood pressure. Atrial natriuretic peptide has important influences on the autonomic nervous system, blocking sympathetic activity even when cardiac filling pressures fall.26 The peptide also causes disruption of the renin–angiotensin–aldosterone axis. In the cortical collecting ducts, the peptide inhibits transport of water by antagonizing the effects of vasopressin.27 In the inner medullary collecting duct, the peptide blocks the absorption of sodium by stimulating production of cyclic guanosine monophosphate.28 Interestingly, transcription of the genes for atrial natriuretic peptide and B-type natriuretic peptide characterizes the growth of cardiac cells, and has been shown to be tightly linked to growth. Such transcription is responsible for proliferation in the fetal heart, and trophic growth in the setting of cardiac hypertrophy.29
The B-type natriuretic peptide
The B-type natriuretic peptide is a cardiac neurohormone synthesized in the cardiac ventricles. It is synthesized as a 134-amino-acid peptide, named prepro-B-type natriuretic peptide. The prepro-peptide is cleaved into pro-B-type natriuretic peptide and a 26-amino-acid sequence signal peptide. The 108 pro-B-type natriuretic peptide sequence is cleaved by furin to form active B-type natriuretic peptide77–108 and inactive N-terminal pro-B-type natriuretic peptide1–76 protein.30 The half-life of N-terminal pro-B-type natriuretic peptide is around 120 minutes and the half-life of B-type natriuretic peptide is around 22 minutes. Prior studies have suggested that the B-type peptide can accurately reflect changes in pulmonary capillary wedge pressure as rapidly as every 2 hours. The peptide was first identified in extracts from the porcine brain. It is also present in the human brain, but in much lower concentrations than in the human ventricle. It has been demonstrated in the human genome that the gene for B-type natriuretic peptide is located on chromosome 1, organized in tandem with the gene for atrial natriuretic peptide.
The B-type peptide has cardiovascular and renal effects that are very similar to those of atrial natriuretic peptide. It causes a decrease in blood pressure by reducing preload secondary to a shift of intravascular fluid into the extravascular compartment. It also increases venous capacitance, which helps to facilitate a natriuresis that reduces the overall extracellular fluid volume. This effect is mediated by direct effects on the renal system, and suppression of the renin–angiotensin–aldosterone axis.31 It reduces sympathetic tone in the peripheral vasculature. This probably occurs by suppression of the release of catecholamines, suppression of sympathetic outflow from the central nervous system, and dampening of baroreceptors.32 The activation threshold of vagal afferents can be reduced by the peptide, thereby suppressing reflex tachycardia and vasoconstriction.
B-type natriuretic peptide, as well as atrial natriuretic peptide and C-type natriuretic peptide, are produced in the brain. The B-type peptide that is produced elsewhere, however, does not cross the blood–brain barrier. Systemic B-type peptide circulating in the plasma does reach sites outside the barrier to the central nervous system barrier, such as the subfornical region, the area postrema, and the hypothalamic median eminence. The actions of the B-type peptide produced locally in the brain reinforce the actions occurring in the other parts of the body. For example, effects of the B-type peptide circulating in the plasma are augmented by the inhibition by the central nervous system of ingestion of water and craving for salt.33
The clinical applications of the B-type peptide are numerous, and rapidly expanding. Elevation of the peptide in the setting of heart failure has been shown to be directly related to morbidity and mortality.34–36 Its measurement has also been shown to be useful in differentiating pulmonary from cardiac aetiologies, such as decompensated heart failure in patients presenting with dyspnea (Fig. 3).37 Levels of the B-type peptide predict outcomes in patients with acute myocardial infarction.38 In contrast, when measured between 1 and 4 days after a transmural infarction, elevated levels have been shown to be associated with increased risk of death independent of left ventricular function.39 Elevated levels of the peptide have also been evaluated in the setting of non-ST-segment elevation myocardial infarction, and found to be important in identifying patients at higher risk of death and heart failure.40 Myocardial ischemia,41 and unstable angina,42 can lead to increased levels of the B-type peptide in the plasma. Levels are increased in many patients with essential hypertension and left ventricular hypertrophy.43 Patients requiring support from ventricular assist devices have been assessed for changes in levels of the B-type peptide, with lower-levels measured after support was instituted.44 Elevated levels have also been noted in patients who have undergone orthotopic heart transplantation. Indeed, measurement of the B-type peptide has been proposed as a biochemical screening tool for detection of left ventricular systolic dysfunction in older adults,45 but this has not been widely accepted.
Figure 3.
BNP concentration in patients with dyspnea. The bars represent the highest and lowest values of BNP measured (nanogram/liter) in patients with no evidence of congestive heart failure vs. dyspnea secondary to non-cardiac causes (but with history of LV dysfunction) and dyspnea secondary to congestive heart failure. (Reproduced with permission from de Lemos JA, et al. B-type natriuretic peptide in cardiovascular disease. The Lancet 2003; 362: 316–322.)
In healthy babies and children, the concentration of the B-type peptide in the plasma increases to a peak immediately after birth, and slowly reaches adult levels by 3 months of age.46 Shortly after birth, the circulatory changes that occur lead to an increase in left ventricular volume, also allow for the synthesis of B-type natriuretic peptide. Plasma N-terminal pro-B-type natriuretic peptide has been evaluated in patients ranging from neonates to young adults, both in healthy populations and those with heart failure. In the healthy subjects, the normal range of N-terminal pro-B-type natriuretic peptide was from 150 and 430 femtomolar per milliliter. When assessed in children with heart failure, the peptide was elevated above these control levels, with the level reflecting the severity of symptoms.47 The B-type peptide has been shown to be elevated in the setting of renal dysfunction. Males without cardiac disease have higher levels in the serum than do females, and levels are also usually higher in the elderly than the middle-aged population without heart disease. There is no evidence of circadian rhythmic variation of the B-type natriuretic peptide.48
Levels of the B-type peptide have been evaluated in specific diseases, such as Duchenne's muscular dystrophy,49 albeit that increases in the peptide did not act as a sensitive marker for the early detection of systolic dysfunction. Detection of elevated levels of the peptide in patients with systolic dysfunction and a decreased deceleration time revealed by echocardiography, nonetheless, suggested a poorer prognosis. The peptide has also been evaluated in the settings of Kawasaki's disease and myocarditis,50, 51 When both the atrial and the B-type peptides were assessed prior to transcatheter closure of atrial septal defects, levels were found to be elevated prior to intervention, but decreased to control levels after successful closure.52
Levels of the B-type peptide in the plasma have also been used as a prognosticator in the setting of primary pulmonary hypertension. Levels above normal, and in particular, an increase of levels during the follow-up of these patients, may be associated with increased mortality.53 Levels of the peptide have also been shown to increase in proportion to the degree of right ventricular dysfunction in the setting of pulmonary hypertension.54
The B-type peptide has been assessed in patients with hypertrophic cardiomyopathy. In this setting, expression at the level of the ventricular myocyte may be augmented in response to both obstruction and diastolic dysfunction.55 In children undergoing doxorubicin therapy for malignant disease, it is postulated that measurements of the levels of the atrial and the B-type peptides in the serum may allow for earlier detection of cardiac damage secondary to the chemotherapeutic regime.56
Only limited data exist in the setting of surgical intervention in patients with congenital cardiac disease. There are reports suggesting that the B-type peptide is elevated in patients with functionally univentricular physiology.57 These studies suggest that the volume overload produced by a palliative operation, such as the Blalock–Taussig shunt, results in increased production of B-type natriuretic peptide. The ventricular unloading that occurs subsequent to a second unloading palliative procedure, such as a Glenn anastomosis, restores the values to the “accepted” normal range. Similar results were seen in patients who underwent total cavopulmonary connection as opposed to repair in the setting of tetralogy of Fallot. Levels of both the atrial and B-type peptides were significantly lower in the early postoperative period in patients converted to the Fontan circulation, suggesting suppression of the release of these hormones in the setting of the total cavopulmonary connection.58 The B-type peptide, as well as aldosterone, has been shown to be increased in the setting of cardiac surgery requiring cardiopulmonary bypass in children, and further shown to decrease from preoperative levels59 following intervention.
The appropriate use of the B-type natriuretic peptide as a valuable diagnostic and clinical tool hinges on the rapidity and sensitivity of its bioassay. A bedside assay for its evaluation was granted approval in the year 2000 by the Food and Drug Administration. This assay was shown to be effective in diagnosing heart failure in patients with acute dyspnea in the Breathing Not Properly Multinational Study.60, 61 Furthermore, a fully automated test for quantification of the N-terminal fragment of N-terminal pro-B-type natriuretic peptide received Food and Drug Administration approval in 2002.
The C-type natriuretic peptide
The third member of the natriuretic peptide family is called C-type natriuretic peptide. This C-type peptide is structurally different from both the atrial and the B-type variants, and is expressed to a larger extent in the central nervous system and other vascular structures than in the heart.62 There have been two separate C-type natriuretic peptide molecules identified, one with 22 amino acids and the other with 53 amino acids. Both are derived from the single pro-C-type precursor.
As previously stated, it is the B-type receptor that binds the C-type natriuretic peptide. This B-type receptor is found throughout the central nervous system,63 albeit predominating in the region of the hypothalamus, and other rostral regions of the brain. This is the area where natriuretic peptides inhibit secretion of arginine vasopressin, and stimulate sympathetic tone.
In contrast with the atrial and B-type peptides, the C-type peptide does not appear to have significant function as a circulating hormone. It acts locally in the vasculature as a vasodilatory agent and inhibitor of vascular cell proliferation, as well as in the central nervous system, where it has numerous functions.64 Its levels have been measured in the setting of heart failure, and compared to healthy controls, and found to be no different.65
Cytokines
The cardiovascular system responds to injury by activation of cytokines in a fashion similar to other human systems of organs. Cytokines are low-molecular-weight proteins that are secreted by several different cells. They have a variety of immune and/or inflammatory functions. The main cytokines associated with roles in the pathophysiology of heart failure are endothelin-1, tumor necrosis factor, and interleukin-6. Elevated levels are associated with a poor prognosis, and are known to have adverse effects on the myocardial structure and function.66
Kinins
Kinin peptides are potent vasodilators, and have a broad spectrum of activity, including the promotion of diuresis and natriuresis. Kinins also protect against ischemia–reperfusion injury by decreasing endothelial adherence of leukocytes. This leads to attenuation of disruption of the microvascular barrier, attenuation of postischemic leukocyte adherence, and reduced patterns of injury.67 In humans, the plasma and tissue kallikrein–kinin system generates bradykinin and kallidin peptides, respectively. These kinins act via two receptors: type 1, also known as B1; and type 2, known as B2, with the latter being the predominant type. Kinins appear to mediate the hypotensive effects of treatment with angiotensin-converting enzyme. They also play an important role in neutrophil chemotaxis, pain, inflammation, vasodilation, and vascular permeability.
Endothelin-1
The endothelins are peptides released from endothelial cells, and have potent vasoconstrictor properties. Endothelin-1 is one such potent cardiovascular peptide that has been shown to cause retention of sodium, vasoconstriction, and mitogenesis. The actions of endothelin-1 are mediated through the A and B receptors. The A receptors are located on vascular smooth muscles, while the B receptors are located on the endothelium. Endothelin-1 is elevated in the setting of congestive heart failure. Its main source in this setting is the pulmonary vascular bed, which may play a role in mediating elevation of cardiac filling pressures and progression of circulatory failure. The production of tumor necrosis factor-alpha can be stimulated by endothelin-1.68 High levels of endothelin-1 are associated with increased mortality in patients with heart failure.69 Endothelin-1 may also play a role in the progression of pulmonary hypertension in heart failure. Accordingly, the severity of pulmonary hypertension is reduced by antagonists to the endothelin receptors.70 These agents have also resulted in improvement in left ventricular performance, and provide a reduction in systemic vascular resistance in patients with heart failure.71 It has been postulated that part of the natriuretic effects of the natriuretic peptides as described above may be due to the suppression of production of endothelin-1.72
Tumor necrosis factor
Tumor necrosis factor-alpha, also known as cachectin, is central to the systemic inflammatory response. This factor results in depression of myocardial function, with administration intravenously of blockers of the factor shown to improve cardiac function in adults with sepsis.73 The factor has also been shown to increase activity of nitric oxide synthase in vascular smooth muscle. Levels of the alpha factor in the serum have also been shown to be elevated in the setting of heart failure, with the increase in levels correlating with the severity of disease.74 Tumor necrosis factor-alpha is produced in the failing heart, and has known negative inotropic effects. It has been found to cause changes in the myocardium, including interstitial fibrosis, ventricular remodeling, and apoptosis.75 Patients with high levels of the factor are more cachectic, have more advanced heart failure, and may develop a wasting syndrome known as cardiac cachexia, which is associated with a poorer prognosis.76 The factor is capable of inducing wasting of skeletal muscles and apoptosis, and can function as an independent predictor of depressed variability of heart rate in patients known to be in heart failure.77 There are two receptors for the alpha factor, known as receptors 1 and 2. Both receptors can be detected as soluble forms. There is a strong correlation between elevated levels of the receptors and 30-day mortality in patients with severe heart failure.78 Treatment with a blocker of the alpha factor known as etanercept has been shown to lead to a dose-dependent improvement in left ventricular ejection fraction and left ventricular remodeling, with a possible trend toward improvement in functional status.79
Vasopressin
Arginine vasopressin is a hormone secreted by the pituitary gland that plays a role in plasma osmolality and clearance of free water. Circulating levels are elevated in the setting of heart failure, and control of these circulating levels can be abnormal in patients with heart failure, especially in those who do not decrease levels with a documented reduction of serum osmolality.80 Patients with heart failure typically lack the normal suppression of release of vasopressin after administration of ethanol.81 Two types of receptors have been identified, and named V1 and V2. In animal studies, the selective blockade of the V1 receptor resulted in an increase in cardiac output without any significant change in levels of hormones or electrolytes. The selective blockade of the V2 receptor resulted in increased levels of vasopressin and sodium, and increased activity of renin, albeit without changes in cardiac output. When both receptors were blocked, the hemodynamic effects were potentiated. These findings suggest that vasopressin not only regulates clearance of free water, but also may contribute through its V1 receptor to the vasoconstriction seen in heart failure.82
Interleukins
The interleukins also play an important role in the pathogenesis of heart failure.83 These molecules are inflammatory cytokines, and have been studied extensively in adults. Interleukin-1 is produced by a variety of cells, and plays an important role in the systemic immune response. It has been shown to depress myocardial contractility by stimulating nitric oxide synthase.84 In addition, it inhibits the beta-adrenergic responsiveness of cardiac myocytes.85 Elevated levels are found in the setting of heart failure, and are associated in adults with a worse standing in the categorization of the New York Heart Association, increased length of hospital stays for decompensated heart failure, and poorer left ventricular function.74
Pharmacologic therapy and biomarkers
The implications of neurochemical monitoring must also be assessed from the perspective of suppression. The Valsartan Heart Failure Trial86 evaluated B-type natriuretic peptide and norepinephrine. Both peptides were measured at baseline and at 4, 12, and 24 months after randomization with either Valsartan, a blocker of the angiotensin-II receptor, or placebo. Valsartan caused sustained reduction in levels of B-type natriuretic peptide, and attenuated increases in norepinephrine, over the time period of the study. This correlated with clinical effects of decreased combined morbidity and mortality, improvement in symptoms, left ventricular function and size, and reduced hospitalizations secondary to heart failure. The second Valsartan Heart Failure Trial demonstrated that treatment with enalapril reduced the levels of norepinephrine in the plasma compared with a control group treated with isosorbide dinitrate and hydralazine.8 The Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure showed favorable long-term effects with the use of bisoprolol and metoprolol, respectively.87 This was also further delineated by Packer et al. in their evaluation using the beta-blocker carvedilol.88 All these studies documented decreases in markers of activation of the sympathetic nervous system via drug therapy, resulting in improvement in long-term morbidity and mortality.
Infusions of synthetic B-type natriuretic peptide have been shown to be beneficial in the treatment of decompensated heart failure. Nesiritide, an intravenous recombinant form of human B-type natriuretic peptide, and marketed as Natrecor by Scios, Incorporated, Fremont, California, is the first in a new pharmacologic class of drug for treatment of decompensated congestive heart failure. Beneficial effects including vascular dilation, suppression of the renin–angiotensin–aldosterone and sympathetic nervous systems, and enhanced excretion of sodium89 have all been observed. The investigators of the Nesiritide Study Group evaluated the efficacy of infusion of the synthetic B-type natriuretic peptide, as well as comparing the use of nesiritide with standard intravenous agents in a controlled design.90 Infusion of the synthetic B-type peptide resulted in dose-dependent decreases in right atrial pressure, systolic blood pressure, systemic vascular resistance, and pulmonary capillary wedge pressure, without causing a significant change in heart rate. Overall clinical state, including dyspnea and fatigue, as assessed by the patient was improved in about two-thirds of the patients. There was also a statistically significant improvement in the global state of the patients as judged by the physicians, in addition to a statistically significant decrease noted of levels of aldosterone in the plasma. Urinary output was also more improved in those receiving the synthetic peptide over the duration of the study. In adults with a history of chronic heart failure, and a need for intravenous therapy of decompensated heart failure, the use of nesiritide in both high- and low-dose infusion resulted in a lower mortality at 6 months, and a trend toward decreased readmissions.91 Its use when compared to dobutamine has been evaluated in the treatment of acutely decompensated heart failure. Dobutamine had substantial chronotropic and pro-arrhythmic effects, whereas nesiritide reduced ventricular ectopy. Finally, in the Vasodilation in the Management of Acute Congestive Heart Failure Trial, intravenous infusion of the synthetic B-type peptide was compared to both placebo and intravenous nitroglycerin in patients with decompensated heart failure. It was concluded that, as measured by pulmonary capillary wedge pressure over periods of 3 and 24 hours, nesiritide improved hemodynamics more effectively than did nitroglycerin.92
In the past, blockers of aldosterone have also been evaluated. In the Randomized Aldactone Evaluation Study, the addition of spironolactone reduced mortality by one-third in patients with heart failure graded at classes III and IV in the categorization of the New York Heart Association, who were already being treated with diuretics, digoxin, and inhibitors of angiotensin-converting enzyme.93 The mechanism resulting in these improved outcomes has not been fully elucidated. Newer blocking agents are now being developed. The most notable is eplerenone. This is an agent that selectively blocks the mineralocorticoid receptor but not the glucocorticoid, progesterone, or androgen receptors. The Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study evaluated its use against placebo in patients with heart failure receiving optimal medical therapy.94 The addition of eplerenone reduced morbidity and mortality among patients with left ventricular dysfunction and heart failure in the setting of myocardial infarction.
Antagonism to vasopressin has also been postulated as a possible treatment for chronic heart failure. The A Dose evaluation of a Vasopressin Antagonist in Congestive Heart Failure Trial evaluated patients using conivaptan, a vasopressin antagonist, assessing its functional capacity in heart failure.95 There are other clinical trials underway currently that are evaluating the utility of such antagonist therapy.95, 96 Their implications for management remain to be delineated.
Current state and future trends
There has been limited reported use in pediatrics. Feingold and Law recently published use of nesitiride in four cases in three subjects with chronic heart failure of various causes with improvement in symptoms and diuresis.97 Smith and colleagues reported the use of nesitiride in two infants on extracorporeal membrane oxygenation for blood pressure control and augmentation of urine output.98 We have recently completed a study at the Texas Children's Hospital in which patients aged 2 to 21 years, and in classes III to IV of the New York Heart Association for grading of heart failure, were enrolled. The results of this study have been submitted for publication, however, during the course of administration of nesitiride, no adverse events have been noted in this population.
Levels of angiotensin-II, renin, aldosterone, vasopressin, atrial natriuretic peptide, and B-type natriuretic peptide have been shown to be elevated in the plasma of patients who had successfully undergone total cavopulmonary connection and bidirectional Glenn procedures. These levels were found to be elevated for years after the surgical procedures, but lower levels were observed over time.99 An abnormal exercise capacity, and elevated neurohormonal activity, has been documented in patients after the Fontan operation.100, 101 A study by Ohuchi et al.102 has evaluated cardiac autonomic nervous activity in this population. They concluded that, in addition to damage secondary to the procedure, the Fontan circulation may impair cardiac autonomic activity.
Levels of the atrial and B-type peptides have also been measured in patients who have undergone complete surgical repair of tetralogy of Fallot receiving dobutamine. Levels were noted to be elevated prior to the infusion, but showed a decrease in the majority of patients after infusion, correlating with right-sided pressure and volume overload.103
The future of neurohormones as markers, and as therapeutic modalities, remains a work in progress as far as children are concerned. More data is being reported, but the amount of information currently available is limited. Much of the information presented in this review is extrapolated from experience with adults. Caution must therefore be used in transposing this data derived from adults to children in heart failure.104
Conclusions
It must be remembered that neurohormonal markers are just that, markers. It is probably unrealistic to expect a single biomarker to offer absolutes in the management of patients in heart failure, much less than to serve as a surrogate for the efficacy of clinical treatment. It is not unreasonable, however, to think that the use of multiple markers can markedly change the face of current strategies for clinical management. In the future, use of these markers may guide the initial approaches to treatment, further aid in optimization of therapy, or serve as a screening tool for the development of heart failure in children. Until that time, more research will need to performed in children further to delineate the potential influence of these markers. This remains an area that is relatively unexplored, and one which will most likely become more important as evidenced by our growing populations of children and adults in heart failure.