


Significant outcomes
-
∙ There were no significant differences in the cortical lipidome of older control subjects, possessing significant Alzheimer’s disease (AD) pathology, to that of younger controls.
-
∙ Monoacylglycerols and diacylglycerols (DAG) were elevated in the grey matter of the subjects with mild cognitive impairment (MCI) and in the old dementia (OD) cohort, but not in the young dementia cohort, suggesting a common underlying pathophysiology in the MCI and OD groups.
-
∙ Phosphatidylethanolamines, potential metabolic precursors of DAGs, were lower in the MCI, YD, and OD cohorts.
-
∙ While ethanolamine plasmalogens were decreased in the YD and OD groups they were unaltered in the MCI group indicating that alterations in plasmalogen synthesis are unlikely to represent an initiating event in the transition from MCI to frank dementia.
-
∙ Low levels of docosahexaenoic acid (DHA) in the cerebrospinal fluid and grey matter suggest that DHA transport and/or metabolism are altered in AD.
Limitations
-
∙ Our non-targeted lipidomics platform generates only relative and not absolute levels of lipids.
-
∙ The N for the old control group is small and requires further studies to validate our observations.
Introduction
The anticipated expansion of individuals surviving into their 80s and 90s in the coming decades is predicted to substantially increase the prevalence of dementia. Currently, therapies that target dementia are focused on processes of Alzheimer’s disease (AD), especially beta-amyloid (Aβ), because of genetic evidence for its causative role in AD and because abnormalities in Aβ develop relatively early in the course of the disease, at a point at which disease modification therapies are more likely to meet with clinical success (Reference Korczyn1). Unfortunately, these approaches that are effective in amyloid mouse models have failed in the clinic (Reference Castello and Soriano2,Reference Farlow and Brosch3), suggesting that this approach has limited potential for blocking disease initiation and/or progression in AD.
Several studies have suggested that the phenotype of dementia is dependent on age, with histologic features of AD predominant among relatively ‘younger’ elderly subjects, and non-Alzheimer’s pathologies among those who are older (Reference Schneider, Arvanitakis, Bang and Bennett4). Chief among the additional pathologies of the aging brain are vascular brain injury, histologically manifest as large and small vessel sclerosis and microvascular ischaemic injury to brain parenchyma, and white matter lesions, identifiable by magnetic resonance imaging scanning as areas of hyperintensity and histopathologically as myelin pallor (Reference Ganguli and Rodriguez5,Reference Erten-Lyons, Woltjer and Kaye6). However, many cognitively intact individuals have morphologic evidence of disease of this nature (Reference Maarouf, Daugs and Kokjohn7–Reference Erten-Lyons, Woltjer and Dodge10). The picture is further complicated by the observation that elderly subjects usually also harbour at least some burden of the lesions of AD, namely amyloid plaques and neurofibrillary tangles (Reference Schneider, Arvanitakis, Bang and Bennett4,Reference Erten-Lyons, Woltjer and Kaye6–Reference Erten-Lyons, Woltjer and Dodge10).
Given ambiguities in the neuropathological basis of cognitive impairment, we undertook to explore aspects of the biochemical features of brain aging and cognitive impairment, focusing on addressing three questions. First, what biochemical features develop in the grey and white matter of cognitively healthy subjects with progressively advancing age? Second, what are the biochemical hallmarks of dementia in the ‘younger’ versus ‘older’ elderly? Third, which biochemical features are present at relatively early stages of cognitive impairment, and therefore are more likely to contribute aetiologically to cognitive decline?
With these questions in mind we evaluated the cerebrospinal fluid (CSF) and frontal cortex lipidomes of younger controls (YC, <85 years), older controls (OC, >85 years), younger demented (YD, <85 years) subjects, older demented (OD, >85 years) subjects, and mild cognitive impairment (MCI) subjects as determined by the Mini-Mental State Examination. The rationale for this approach was based upon the published observations of alterations in AD brain lipids including decreases in plasmalogens and sulphatides, and increases in ceramides and lysoglycerophospholipids (Reference Wood11). An integrated assessment of AD lipidomics utilising high-resolution mass spectrometric methods is required since decrements in brain plasmalogen levels range from large (Reference Ginsberg, Rafique, Xuereb, Rapoport and Gershfeld12,Reference Han, Holtzman and McKeel13) to small (Reference Kou, Kovacs and Höftberger14,Reference Chan, Oliveira and Cortes15). Similarly, large (Reference Cutler, Kelly and Storie16,Reference Han, Holtzman, McKeel, Kelley and Morris17) and small (Reference Chan, Oliveira and Cortes15) increments in brain ceramides and decrements in sulphatides have been reported.
Materials and methods
Human subjects
Post-mortem tissue and CSF were provided to the Oregon Brain Bank by volunteer subjects who were evaluated at Oregon Health and Science University (IRB 00001623). Subjects were categorised by age and cognitive function described in Table 1. Cognitive impairment was assessed in the Layton Aging and Alzheimer’s Disease Center with annual neurological and neuropsychological evaluations, and clinical dementia rating (CDR) as assigned by an experienced neurologist. Controls had normal examinations. The clinical diagnosis of dementia was determined by clinical team consensus conference. Patients with dementia met the National Institute for Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorder Association diagnostic criteria for clinical AD, and had a CDR>1. All subjects had pathologic diagnoses determined at the Oregon Alzheimer’s Disease Center. Tissue and CSF acquisition and use followed institutional review board requirements. Frontal lobe tissue and CSF were flash frozen and stored at −80°C for biochemical studies described here. Brain tissue was evaluated by a neuropathologist for features of neurodegenerative disease, which were identified and staged as previously reported (Reference Erten-Lyons, Woltjer and Kaye6) and according to the National Alzheimer’s Coordinating Center’s uniform data collection. A modified Bielschowski silver impregnation method was used to identify diffuse and neuritic plaques, a PHF1 antibody (a gift from Dr. Peter Davies, Albert Einstein College of Medicine, Manhasset, NY) was utilised for Tau immunohistochemistry, and the presence of Lewy bodies was determined by anti-α-synuclein immunohistochemistry using LB509 antibody (Covance, Princeton, NJ, USA).
Table 1 Demographic and pathologic data for control, mild cognitive impairment (MCI) and Alzheimer’s disease groups
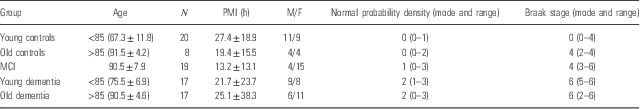
PMI, post-mortem interval.
Lipid analyses
CSF (0.5 ml) and frozen frontal lobe grey and subjacent white matter samples were processed separately utilising tert-butylmethylether and methanol for extraction of lipids (Reference Schuhmann, Almeida, Baumert, Herzog, Bornstein and Shevchenko18–Reference Wood, Filiou, Otte, Zimmer and Turck20). The extraction solution contained [2H8]arachidonic acid, [2H3]phytanic acid, [2H4]hexacosanoic acid, [2H28]hexadecanedioic acid, [13C16]palmitic acid, [2H7]cholesterol sulphate, [2H5]MAG 18:1, [2H3]Carnitine 18:0, [13C3]DAG 36:2, [2H5]TAG 48:0, [2H31]PtdEtn 34:1, [2H54]PtdEtn 28:0, [2H31]PtdCh 34:1, [2H54]PtdCh 28:0, [2H62]PtdCh 32:0, [2H31]SM 16:0, [2H31]PtdSer 36:1, [2H31]PA 34:1, [2H62]PG 32:0, bromocriptine, and glyburide as internal standards. Extracts were dried by centrifugal vacuum evaporation before dissolution in isopropanol : methanol :chloroform (4 : 2 : 1) containing 7.5 mM ammonium acetate. Non-targeted lipidomics were performed utilising high-resolution (140000 at 200 amu) data acquisition, with sub-millimass accuracy on an orbitrap mass spectrometer (Thermo Q Exactive, Waltham, Mass), with successive switching between polarity modes (Reference Schuhmann, Almeida, Baumert, Herzog, Bornstein and Shevchenko18–Reference Wood, Filiou, Otte, Zimmer and Turck20). Samples were infused for 2 min at 5 μl/min followed by successive 500 μl washes of the infusion line with methanol and hexane/ethyl acetate (3 : 2) to minimise ghost effects. In negative ion electrospray ionization (ESI) (3.2 kV, capillary temperature of 320°C, sheath gas of 10), the anions of ethanolamine plasmalogens (PlsEtn), phosphatidylglycerols (PG), phosphatidic acids (PA), lysophosphatidic acids, phosphatidylinositols (PtdIn), phosphatidylserines (PtdSer), lysophosphatidylethanolamines (LPE), lysophosphatidylinositols (LPI), lysophosphatidylglycerols (LPG), cerebrosides, sterol sulphates, unsaturated fatty acids, branched-chain fatty acids, and very-long-chain fatty acids (VLCFA), the [M+HCOO]− anions of ceramides, and the [M-2H]− anions of cardiolipins were quantitated and lipid identities validated by MS/MS. In positive ion ESI (4.3 kV, capillary temperature of 320°C, sheath gas of 10), the cations of monoacylglycerols (MAG), choline plasmalogens (PlsCh), phosphatidylcholines (PtdCh), lysophosphatidylcholines (LPC), alkenyl-acyl glycerols, ceramides, ceramide-1-phosphates, and sphingomyelins (SM) and the ammonium adducts of diacylglycerols (DAG), triacylglycerols (TAG), ubiquinone-10, and cholesterol esters were quantitated and lipid identities validated by MS/MS. The anion of bromocriptine and cations of both bromocriptine and glyburide were used as internal standards and to monitor for potential mass axis drift. All ions were monitored with 0.2–3 ppm error.
Data analyses
Data are presented as R values (ratio of the endogenous lipid peak area to the peak area of an appropriate internal standard), corrected for tissue wet weight, in bar graphs±SEM or as box plots. Data were analysed with the Kruskal–Wallis test, followed by the Dunn’s t-test to compare groups to the YC group.
Results
Ethanolamine glycerophospholipids
PlsEtn, particularly those with polyunsaturated fatty acids (PlsEtn 36:4, PlsEtn 38:4, PlsEtn 38:6, PlsEtn 40:4, PlsEtn 40:6) at sn-2, were decreased in the grey matter of the YD and OD groups (Figs 1 and 2). Plasmalogens were not altered in the white matter of the AD cohorts while CSF plasmalogens were very low and extremely variable. Polyunsaturated phosphatidylethanolamines (PtdEtn 36:4, PtdEtn 38:4, PtdEtn 38:6, PtdEtn 40:4) also were decreased in the grey matter of the MCI, YD, and OD groups but only in the white matter of the OD group (Figs 1 and 2). CSF levels of phosphatidylethanolamines were extremely variable and did not differ between cohorts. No changes in LPE levels were noted in grey or white matter (data not shown).
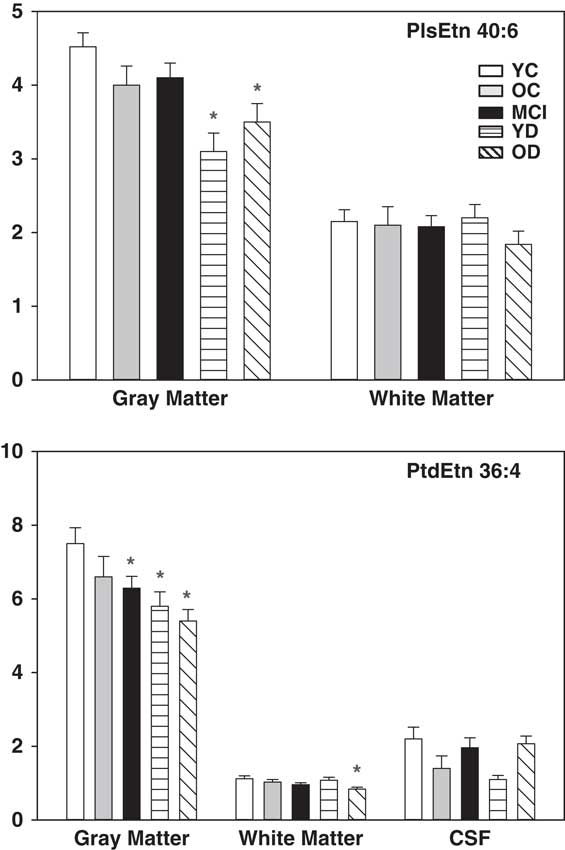
Fig. 1 Levels of ethanolamine plasmalogens (PlsEtn 40:6) and phosphatidylethanolamines (PtdEtn 36:4) in grey matter, white matter, and CSF. X axis=ratio of plasmalogen or phosphatidylethanolamines peak area to the peak area of a stable isotope internal standard, corrected for tissue wet weight. Mean±SEM. *, p<0.05 versus YC. CSF, cerebrospinal fluid; YC, young controls; OC, old controls; MCI, mild cognitive impairment; YD, young dementia; OD, old dementia.
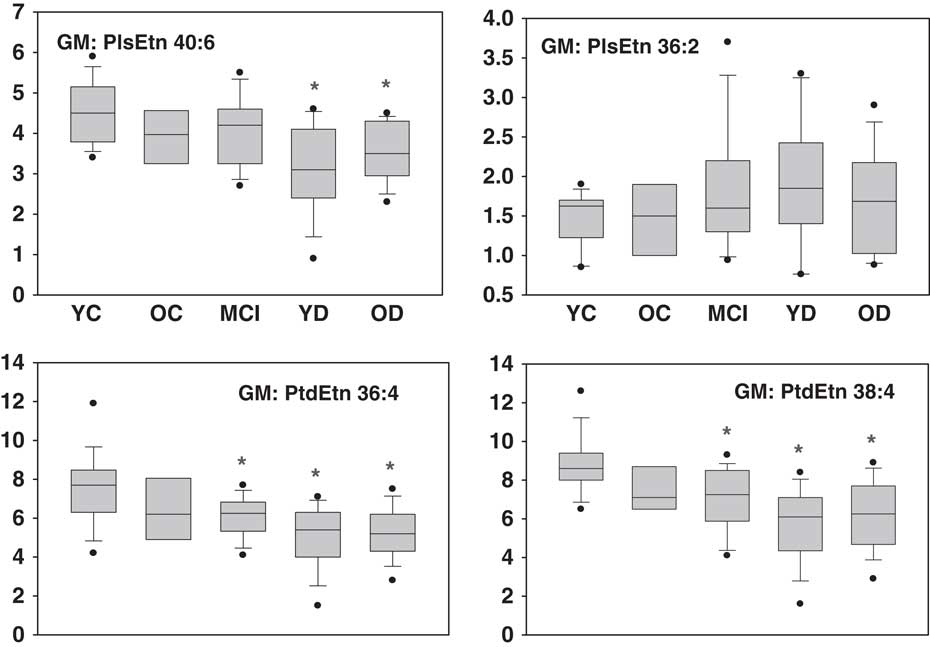
Fig. 2 Box plots of ethanolamine plasmalogen (PlsEtn) and phosphatidylethanolamine (PtdEtn) levels in the grey matter. X axis=ratio of plasmalogen or phosphatidylethanolamines peak area to the peak area of a stable isotope internal standard, corrected for tissue wet weight. Mean±SEM. *, p<0.05 versus YC. YC, young controls; OC, old controls; MCI, mild cognitive impairment; YD, young dementia; OD, old dementia.
Choline glycerophospholipids
Levels of PlsCh, PtdCh, and LPC were unchanged in both the grey and white matter (data not shown). Phosphatidylcholines in the CSF also were not different between groups.
Serine glycerophospholipids
Levels of PtdSer were unaltered in the CSF, grey, or white matter (data not shown).
Inositol glycerophospholipids
Levels of PtdIn were unaltered in the grey and white matter (data not shown).
Glycerol glycerophospholipids
Levels of phosphatidylglycerols and LPG were unaltered in CSF, grey, or white matter (data not shown).
Neutral glycerolipids
Increased MAG (MAG 16:0, MAG 18:0, MAG 18:1) and DAG (DAG 36:1, DAG 36:2, DAG 36:3) levels were noted in the grey matter of the MCI and OD groups, while TAG levels were unaltered (Fig. 3). DAG levels (DAG 34:1, DAG 26:1, DAG 36:2, DAG 36:3) were elevated in the white matter of the MCI group and MAG levels (MAG 16:0, MAG 18:0) were increased in the white matter of the YD and OD groups (Fig. 3). In the MCI group, the white matter MAG levels were heterogeneous with 8 of the 19 subjects having normal MAG levels while the other subjects had three- to six-fold elevations of MAGs. DAG and MAG were not reliably quantitated in the CSF.
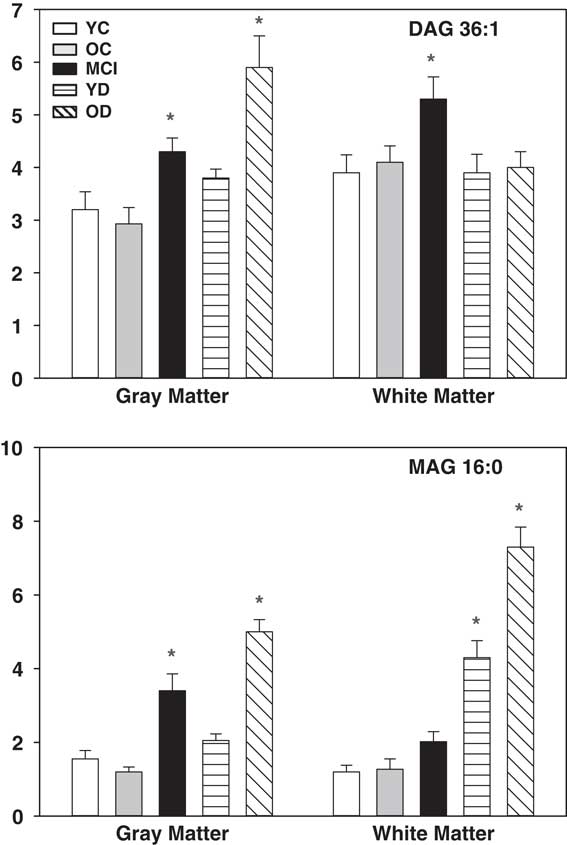
Fig. 3 Levels of DAG (DAG 36:1) and MAG (MAG 16:0) in grey and white matter. X axis=ratio of DAG or MAG peak area to the peak area of a stable isotope internal standard, corrected for tissue wet weight. Mean±SEM. *, p<0.05 versus YC. DAG, diacylglycerols; MAG, monoacylglycerols; YC, young controls; OC, old controls; MCI, mild cognitive impairment; YD, young dementia; OD, old dementia.
Isoprenoids
Ubiquinone-10 levels were not significantly different between groups in the grey matter (data not shown).
Peroxisomal biomarkers
The VLCFA 26:0 was increased in the grey matter of the MCI, YD, and OD groups (Fig. 3). In contrast, DHA levels were lower in the grey matter of the YD and OD groups and the CSF of the MCI and YD groups (Fig. 3).
Sphingolipids
Levels of ceramides, and SM were unaltered in grey matter, white matter, and CSF (data not shown). Sulphatides [24:1, 24:1(OH), 16:0(OH), 24:0(OH)] were decreased in the white matter of the YD group, but unaltered in grey matter, and were not reliably quantitated in CSF (Fig. 4).
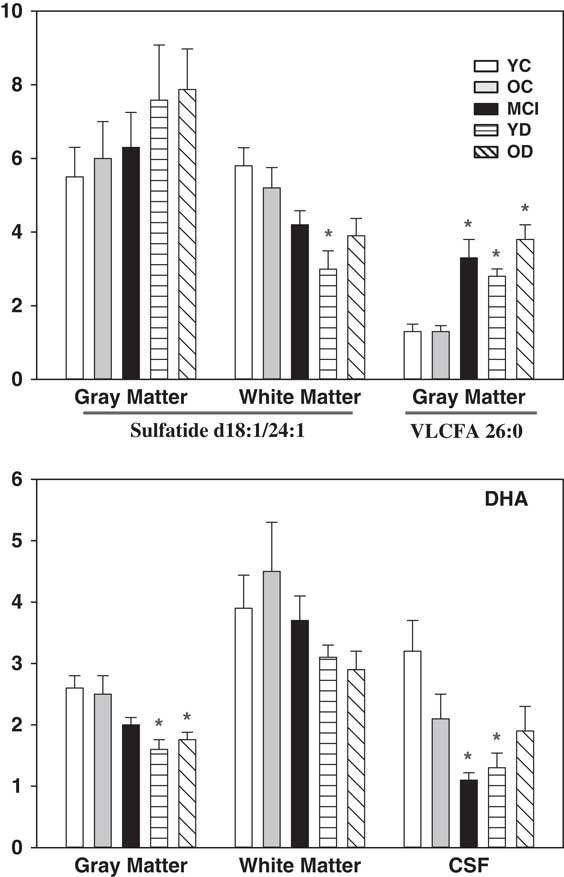
Fig. 4 Levels of the sulphatide (d18:1/24:1), VLCFA 26:0, and DHA. X axis=ratio of lipid peak area to the peak area of a stable isotope internal standard, corrected for tissue wet weight. Mean±SEM. *, p<0.05 versus YC. VLCFA, very-long-chain fatty acid; DHA, docosahexaenoic acid; YC, young controls; OC, old controls; MCI, mild cognitive impairment; YD, young dementia; OD, old dementia.
Summary of negative lipidomics observations
The negative findings with the lipid classes listed above are consistent with previous studies (Reference Han, Holtzman and McKeel13–Reference Cutler, Kelly and Storie16,Reference Han, Holtzman, McKeel, Kelley and Morris17,Reference Cheng, Wang, Li, Cairns and Han21).
Discussion
Previous lipidomics studies of AD have provided an inconsistent picture of overall changes in lipid metabolism. While initial reports of dramatic decrements in brain plasmalogens (Reference Ginsberg, Rafique, Xuereb, Rapoport and Gershfeld12,Reference Han, Holtzman and McKeel13) lead to the subsequent demonstrations of peroxisomal dysfunction in AD brain (Reference Kou, Kovacs and Höftberger14), a number of reports have observed much more modest decrements in brain plasmalogens (Reference Kou, Kovacs and Höftberger14,Reference Chan, Oliveira and Cortes15). Our observations are consistent with modest decrements in plasmalogens in the YD and OD groups, and no decreases in the MCI group supporting previous reports of normal plasmalogen levels in MCI (Reference Cheng, Wang, Li, Cairns and Han21). These data suggest that plasmalogen deficiency is not an early event in the AD disease process but may well contribute to the progression of the later stages of AD since these are essential lipids for membrane function. Peroxisomal dysfunctional was further validated in our study via measurements of VLCFA accumulation in the MCI, YD, and OD groups and of DHA decrements in the YD and OD group, supporting previous reports (Reference Kou, Kovacs and Höftberger14).
A similar disparity exists in the published literature regarding the degree of decrements in sulphatides and increases in ceramides in AD white matter. Large decrements in brain sulphatides and concomitant large increases in ceramides have been reported by several groups (Reference Cutler, Kelly and Storie16,Reference Han, Holtzman, McKeel, Kelley and Morris17,Reference Cheng, Wang, Li, Cairns and Han21), while other investigators have found only modest alterations in these sphingolipids (Reference Chan, Oliveira and Cortes15). Our high-resolution mass spectrometric lipidomics data are consistent with only modest alterations in these sphingolipid pools.
The most interesting observation from our non-targeted lipidomics analysis was that of augmented DAG levels in AD frontal cortex, confirming a previous report (Reference Chan, Oliveira and Cortes15). In addition, our data is the first to demonstrate elevated DAG levels in the frontal cortex of MCI subjects, indicating that this biochemical alteration occurs early in the AD process. The mechanism(s) for augmented DAG levels in the frontal cortex remain to be accurately defined (Fig. 5). TAGs are unlikely to be metabolic precursors to augmented DAG levels since TAG levels are very low in the brain and are generally unaltered in AD cortex (Reference Chan, Oliveira and Cortes15 and current study). Similarly, the phosphatidic acid pool is an unlikely metabolic source since levels of these lipid metabolites are unaltered in AD cortex (13,Reference Chan, Oliveira and Cortes15 and current study). PtdEtn may be a source via phospholipase C (PLC) since the levels of a number of PtdEtn species are lower in the frontal cortex of AD samples (Reference Chan, Oliveira and Cortes15 and current study). In this regard, increased levels of several PLC isoforms have been detected in AD brain (Reference Shimohama, Sasaki and Fujimoto22,Reference Zhang, Dhillon, Prasad and Markesbery23). However, since PtdSer, PtdCh, PtdIn, and Ptdglycerol levels are unaltered in AD or MCI cortex (13,Reference Chan, Oliveira and Cortes15 and current study), the potential metabolism of PtdEtn by a specific PLC isoform, to generate DAGs, requires further investigation (Fig. 5).
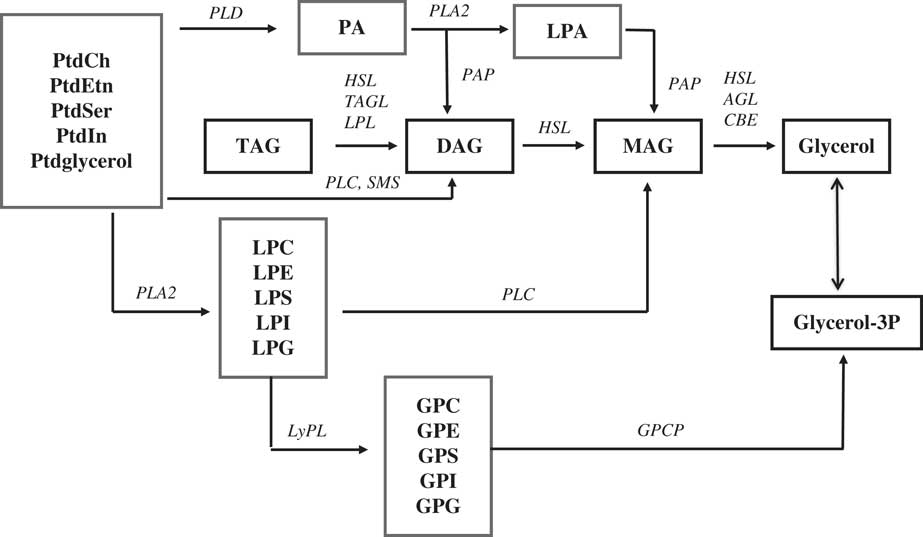
Fig. 5 Schematic presentation of the metabolic sources of diacylglycerols (DAG) and monoacylglycerols (MAG) and their ultimate metabolism to glycerol. AGL, alkyglycerol lipase/MAG lipase (EC 3.1.1.23); CBE, carboxylesterase (EC 3.1.1.1); GPCP, glycerophosphocholine phosphohydrolase (EC 3.1.4.2); HSL, hormone sensitive lipase (EC 3.1.1.79); LPL, lipoprotein lipase/DAG lipase (EC 3.1.134); LyPL, lysophospholipase (EC 3.1.1.5); PAP, phosphatidic acid phosphatase (EC 3.1.3.4); PLA2, phospholipase A2 (EC 3.1.1.4); PLC, phospholipase C (EC 3.1.4.11); PLD, phospholipase D (EC 3.1.4.4); TAGL, triglyceride lipase (EC 3.1.1.3).
The findings of altered DAG levels in MCI and AD brain have recently been extended to plasma studies. We were the first to demonstrate elevated DAG levels in the plasma of MCI and AD patients (Reference Wood, Phillipps, Woltjer, Kaye and Quinn24) and the observations in AD patients have been replicated by another group (Reference González-Domínguez, García-Barrera and Gómez-Ariza25). These data suggest that elevations in DAG levels may represent a broad metabolic alteration in MCI and AD patients.
In light of the diverse structural and signal transduction roles of DAG pools, these are important new observations of a critical biochemical change that we have demonstrated occurs early in the pathophysiology of AD. Brain DAG pools are normally tightly regulated since these glycerolipids serve multiple critical roles. These roles include (Reference Carrasco and Mérida26): (i) precursors for structural glycerophospholipids, TAGs, and MAGs; (ii) mediators of signal transduction via activation of protein kinases, chimaerins, and munc13 proteins; (iii) regulation immune function; and (iv) structural roles in the Golgi, endoplasmic reticulum, and nuclear envelope. Clearly, altered DAG levels could lead to significant structural and signal transduction alterations that could contribute to the deterioration of cognitive brain circuits in AD.
Our data also are the first to demonstrate increases in MAG that parallel increases in DAG levels in AD frontal cortex. PLC hydrolysis of lysoglycerophospholipids might be a metabolic source for MAG, but levels of LPC, LPE, lysophosphatidylserine, LPG, LPI were unaltered in AD cortex (Reference Chan, Oliveira and Cortes15 and current study). Hydrolysis of augmented DAG levels is a more likely source (Fig. 5). In this regard, the protein levels of DAG lipase are augmented in AD hippocampus (Reference Farooqui, Liss and Horrocks27,Reference Mulder, Zilberter and Pasquaré28). Unfortunately, DAG lipase has not been evaluated in AD frontal cortex. Increased protein levels of MAG lipase have also been detected in AD hippocampus (Reference Mulder, Zilberter and Pasquaré28). If these lipase enzyme activities are also augmented in AD frontal cortex, the generation of MAGs must be much faster than subsequent degradation of MAG pools by MAG lipase.
The impact of augmented pools of MAGs in AD remains to be determined. However, we know that the MAG, 2-arachidonyl glycerol, is an endocannabinoid that has been hypothesised to contribute to synaptic failure in AD (Reference Mulder, Zilberter and Pasquaré28). Since MAG lipase is a presynaptic enzyme in glutamatergic neurons (Reference Gulyas, Cravatt and Bracey29) and is present in microglia (Reference Savinainen, Saario and Laitinen30), the increased levels of MAG we monitored in AD frontal cortex may represent biomarkers of glutamate dysfunction (Reference Paula-Lima, Brito-Moreira and Ferreira31) and microgliosis (Reference Wood32), respectively in AD.
With regard to fatty acid alterations in AD, augmented levels of VLCFA and decrements in DHA have been reported (Reference Wood11), observations that we replicated. Accumulation of VLCFA may well contribute to neuronal compromise as suggested previously (Reference Anderson and Stahl33). The lower levels of DHA are complex in that decrements can involve decreased dietary intake, decreased transport, decreased synthesis by brain peroxisomes, and altered metabolism. Since dramatically augmenting the dietary supply of DHA to AD patients appears to have no impact on cognition (Reference Quinn, Raman and Thomas34), it will be important to increase our understanding of the transport of DHA between brain compartments and DHA dynamics in lipid remodelling in AD (Reference Wood11).
Before drawing conclusions from our data it is important to briefly review the study limitations. First, the N of the old control group was only eight potentially biasing our data in this group. However, this was an important group to include since these subjects represent what has been termed non-demented AD neuropathology (NDAN), patients with no cognitive deficit but AD neuropathology at autopsy (Reference Maarouf, Daugs and Kokjohn7,Reference Bjorklund, Reese, Sadagoparamanujam, Ghirardi, Woltjer and Taglialatela35,Reference Wood and Wood36). It is interesting to note that despite the presence of plaques and tangles in these subjects there were none of the lipid alterations that we observed in MCI, YD or OD subjects. These findings further validate the specificity of the findings of elevated DAGs in MCI and OD subjects and suggest that this lipid pool may contribute in the transition from MCI to OD. Second, while we obtained a snapshot of the steady-state levels of a large array of lipids in the brain lipidome, these data in no way assess the potential dynamics of pools that might have altered turnover rates in AD but unaltered steady-state levels. Third, there was a wide range of post-mortem intervals in the subject groups. However, analysis of the data did not detect any correlation between post-mortem interval and lipid levels. These findings agree with our previous analysis of the effects of post-mortem interval on glycerophospholipids and sphingolipids in human skeletal muscle, which revealed that these structural lipids begin to deteriorate at 5–10 days post-mortem (Reference Wood and Shirley19).
Conclusion
In summary, our data support a deficit in PlsEtn in AD but demonstrate that these deficits are not present in MCI, indicating that this is not an early component in the pathogenesis of AD. The accumulation of VLCFA and decrements in DHA support a peroxisomal and/or lipid transport defect that results in the accumulation of toxic VLCFA and shortage of DHA for lipid remodelling. The major new findings of our study were the observations of augmented MAGs and DAGs in the grey matter of the MCI and OD cohorts, suggesting that these biochemical changes may play a role development of MCI and in the transition from MCI to OD. Parallel decrements in phosphatidylethanolamines suggest that this may be a metabolic source for augmented DAGs in MCI and OD, potentially via augmented PLC activity. Since DAGs play multiple structural roles in the brain and both MAGs and DAGs are mediators of signal transduction, it will be important to further evaluate the role(s) of these lipids in the pathogenesis and or progression of AD.
Acknowledgements
The authors thank the participants of the aging study and their families for their valuable contribution to aging and Alzheimer’s research. Authors’ contributions: All authors contributed to the design of the experiment, analysis of the data, and writing the manuscript. R.L.W. performed the neuropathology and P.L.W. performed the lipidomics studies.
Financial Support
This research was funded by Lincoln Memorial University, DeBusk College of Osteopathic medicine, and by NIA-AG08017.
Conflicts of Interest
The authors declare no conflicts of interests.
Ethical Standards
Tissue and post-mortem CSF were provided to the Oregon Brain Bank by volunteer subjects who gave informed consent. The study was approved by the Oregon Health and Science University Institutional Review Board. Written informed consent was obtained from all participants of the Oregon Health and Science University aging study.






