Introduction
Avian haemosporidian infections are caused by Plasmodium malaria parasites, but also by evolutionary closely related ‘malaria-like’ parasites Haemoproteus belonging to Apicomplexa protozoans, order Haemosporida (Pérez-Tris et al., Reference Pérez-Tris, Hasselquist, Hellgren, Krizanauskiene, Waldenström and Bensch2005). These parasites infect the organs and blood of avian hosts (Valkiūnas, Reference Valkiūnas2005), and are transmitted by dipteran vectors (Santiago-Alarcón et al., Reference Santiago-Alarcón, Palinauskas and Schaefer2012). Despite intensive research during the last few decades, understanding of ecological and physiological factors related to the acquisition of infection, as well as its consequences in both hosts and vectors, is still fragmentary. For example, parasitaemia may increase with vector abundance, but not necessarily because of novel parasite transmissions, but as a plastic response of parasites to increased probability of transmission to other hosts (e.g. Cornet et al., Reference Cornet, Nicot, Rivero and Gandon2014). In the same way, effects of infection on host fitness need detailed studies, especially in wild populations (e.g. Knowles et al., Reference Knowles, Wood, Alves, Wilkin, Bensch and Sheldon2011; Asghar et al., Reference Asghar, Hasselquist, Hansson, Zehtindjiev, Westerdahl and Bensch2015; Coon et al., Reference Coon, García-Longoria, Martin, Magallanes, de Lope and Marzal2016). Thus, it is important to keep identifying ‘model organisms’ that may be easily studied in the wild, to reach a deeper understanding of avian haemosporidian infections, especially in the tropics.
The rufous-collared sparrow, Zonotrichia capensis, has a broad geographic distribution from southern Mexico to Patagonia, Argentina, including non-Amazonian Brazil, the Guianas and Venezuela, occupying open spaces, from sea-level to high elevation (Rising and Jaramillo, Reference Rising, Jaramillo, del Hoyo, Elliott, Sargatal, Christie and de Juana2017). In Ecuador, it is common along the Andes, at 1500–3500 m a.s.l. (Ridgely and Greenfield, Reference Ridgely and Greenfield2001). Prevalence of haemosporidian infections in this host has been reported to reach up to 42% of local populations at mid altitude (2000–3300 m a.s.l.) in Andean forests of western Peru (Jones et al., Reference Jones, Cheviron and Carling2013) and up to 73% in the foothills of the Ecuadorian Amazon (1500 m a.s.l.; Escallón et al., Reference Escallón, Weinstein, Tallant, Wojtenek, Rodríguez-Saltos, Bonaccorso and Moore2016). Such high prevalence has been associated with higher major histocompatibility complex diversity, suggesting important parasite-mediated selection on these genes central in the immune response (Jones et al., Reference Jones, Cheviron and Carling2014).
Given the high prevalence of haemosporidian infections reported for rufous-collared sparrows and the implications that it may have for understanding infection dynamics and host resilience, we focused our study on a population of this species in an underrepresented and relatively simple ecosystem: the Andean dry forest. To obtain a first approximation to the system we: (1) identified molecular lineages and morphospecies of Haemosporida parasites infecting this host, as well as their prevalence and parasitaemia; (2) assessed differences in prevalence and parasitaemia caused by different parasites and (3) analysed biotic and abiotic factors associated with infection status and parasitaemia. Based on the relationship between water availability and vector abundance, we hypothesize that both parasitaemia and prevalence of infection would be highly dependent on precipitation in this dry forest ecosystem. Additionally, low diversity of parasite lineages would be expected given a relative low diversity of potential hosts found previously in the study area (Cadena-Ortiz et al., Reference Cadena-Ortiz, Varela, Bahamonde-Vinueza, Freile and Bonaccorso2015), and infection status (infected vs non-infected) by different parasite lineages should have different effects on host body condition.
Materials and methods
Study area and field methods
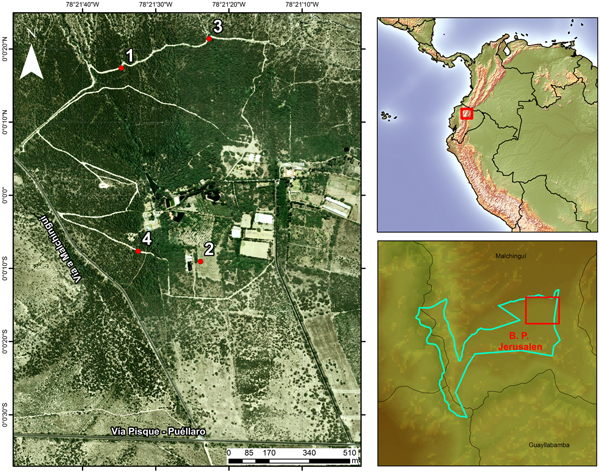
Fieldwork was conducted at Bosque Protector Jerusalem (BPJ), a 1110-ha protected tropical dry forest, ~10 km north of Quito (00°00′05″N, 78°21′18″W), in the Andean valley of Guayllabamba, Ecuador (Fig. 1). The dry season takes place from May to August (125 mm rainfall average), while the wet season occurs from September to April (360 mm rainfall average); average annual temperature under shade is 19 °C (data provided by Instituto Nacional de Meteorología e Hidrología, INAMHI). The study area is mostly covered by ‘semi-deciduous forest and scrub’, characteristic of the northern portion of the inter-Andean valleys of Ecuador (MAE, 2013). This ecosystem is dominated by algarrobo trees (Acacia macracantha), upon which abundant epiphytic vegetation develops, as well as cacti of various genera (e.g. Opuntia sp. and Cleistocactus sp.).
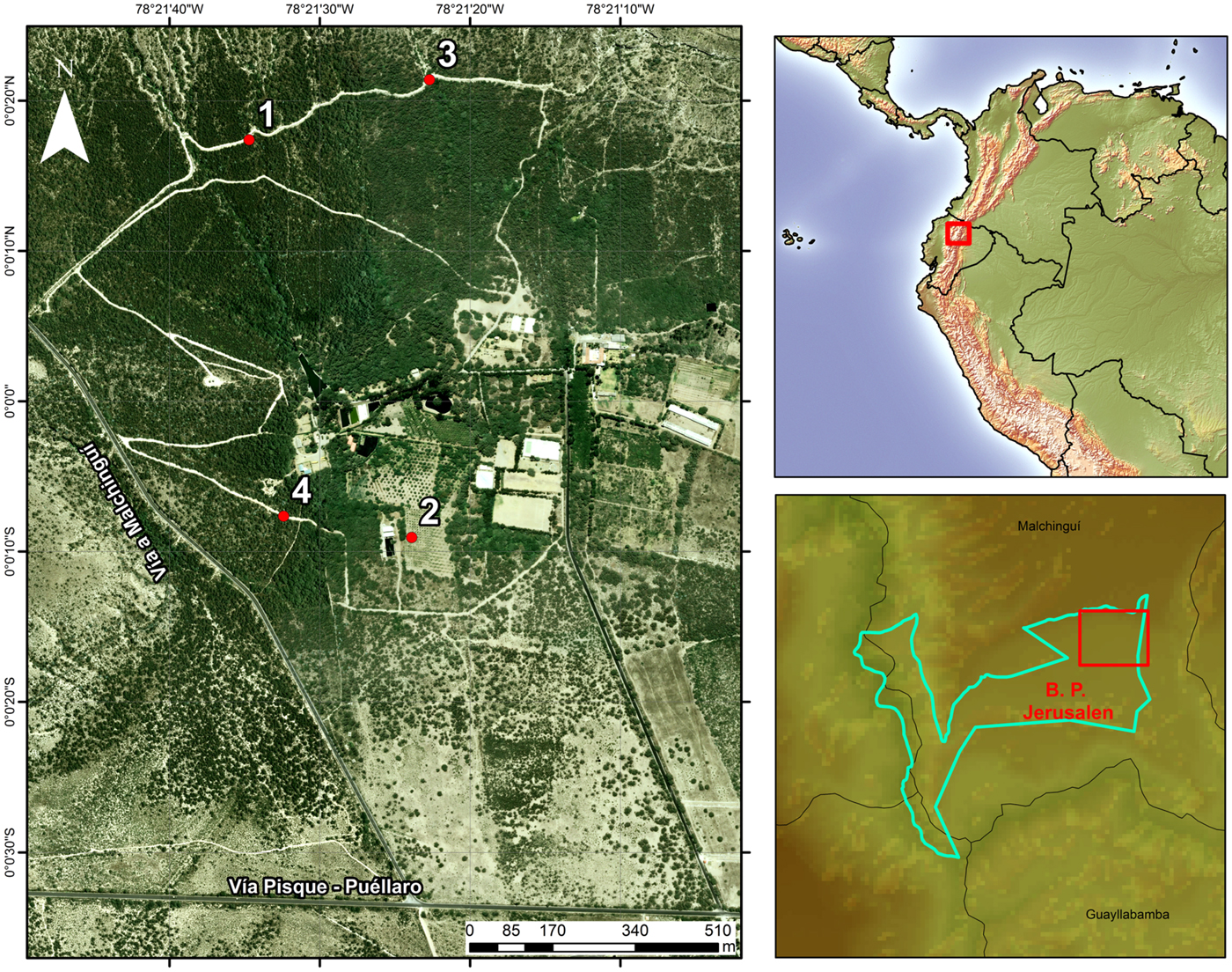
Fig. 1. Study area: sampling sites within Bosque Protector Jerusalem and location of Bosque Protector Jerusalem.
Mist-netting was carried out between December 2012 and June 2013, at four sampling sites that aimed to account for the environmental heterogeneity of the reserve (Fig. 1). Sites 1 and 3 were located over a trail within the ‘core’ of the dry forest, and were separated by 320 m. These two sites are bordered on both sides by dense, native vegetation. There, the canopy is dominated by Acacia sp. (<4 m), harbouring a great variety of epiphytes, and Cactaceae are abundant in the understory. Sites 2 and 4 were established on a more disturbed area of the reserve and separated by 200 m. Site 2 was located within a plantation of Citrus sp. and Persea americana (<3 m); along the borders of this plantation, remnant patches of native vegetation still exist, alternated by introduced trees of Eucalyptus sp. (<10 m). Site 4 was located on a path within the dry forest, where native vegetation exists but in lower density than on sites 1 and 3. Sites 1 and 3 were separated from sites 2 and 4 by more than 600 m. Each site was visited in sequential order (1–4), conducting a total of six visits per site (1.5-day visit per month, per site). At each site, we placed seven mist-nets (four shelves, 12 m × 2.5 m; 36 mm mesh). Nets were opened from 6:00 to 18:00 h the first day, and from 6:00 to 12:00 the second day. All birds captured were weighed to the nearest 0.1 g, and measured to the nearest 0.01 mm (bill, tarsus) or the nearest millimetre (wing). We obtained blood samples from the jugular or brachial vein using disposable needles; 5–20 µl were used to prepare a blood smear (for microscopic analysis) and 20 µl were preserved in 99% ethanol solution (for molecular analysis). At the end of this process, birds were banded and released. After air drying, smears where fixed with methanol for 3 min in the field and then stained with Giemsa for 60 min in the laboratory (Waldenström et al., Reference Waldenström, Bensch, Hasselquist and Ostman2004).
Molecular lineage identification and phylogenetic relationships
To determine the identity of haemosporidian lineages, as well as their evolutionary relationships, we performed phylogenetic analyses based on partial sequences of the mitochondrial gene cytochrome b (cytb). Total genomic DNA was extracted from blood using a lysis protocol with proteinase K and protein precipitation with guanidine thiocyanate (Chomczynski, Reference Chomczynski1993). DNA amplification was conducted based on a non-nested polymerase chain reaction (PCR) protocol, using primers HaemF and HaemR2 (Waldenström et al., Reference Waldenström, Bensch, Hasselquist and Ostman2004). PCR reactions were prepared in 25 µl volumes with 5 µl of genomic DNA, 3 mm MgCl2, PCR buffer (Invitrogen Inc., Carlsbad, USA), 0.4 mm dNTPs, 0.6 µ m of each primer and 1.25 U Platinum Taq polymerase (Invitrogen), using the PCR profile described in Waldenström et al. (Reference Waldenström, Bensch, Hasselquist and Ostman2004). Final PCR products were stained with SYBR Safe (Invitrogen) and visualized on a 1% agarose gel. When samples tested negative, we used 1 µl of the PCR product, along with the same reagent proportions and thermocycler profile (using 20 cycles) in a second PCR, to try to rule out false negatives. PCR amplicons from positively infected individuals were purified using ExoSAP-IT (Affymetrix Inc., Santa Clara, USA) and sequenced in an ABI 3730XL sequence analyzer (Applied Biosystems Inc., Foster City, USA), with the same primers used for PCR. Chromatograms of parasite cytb were assembled and edited with Geneious 5.1.7 (Kearse et al., Reference Kearse, Moir, Wilson, Stones-Havas, Cheung, Sturrock, Buxton, Cooper, Markowitz, Duran, Thierer, Ashton, Mentjies and Drummond2012).
Sequences depicting single infections were aligned automatically in Clustal X2 (Larkin et al., Reference Larkin, Blackshields, Brown, Chenna, McGettigan, McWilliam, Valentin, Wallace, Wilm, Lopez, Thompson, Gibson and Higgins2007) to identify samples infected with identical lineages. Sequences showing double peaks in chromatograms were treated as coinfections and resolved through computational phasing in the program PHASE (Stephens and Donnelly, Reference Stephens and Donnelly2003) as implemented in DNAsp 5 (Librado and Rozas, Reference Librado and Rozas2009). In the PHASE analysis, we included nine sequences with coinfections and ten additional sequences – three identified during molecular diagnosis (see Results) and seven additional sequences that correspond to the molecular lineages associated with morphospecies found during morphological identification (five Haemoproteus (Parahaemoproteus) coatneyi, one H. (P.) erythrogravidus and one Plasmodium (Novyella) nucleophilum; see Results). The analysis was run with default parameters (except for 1000 iterations, thinning interval = 10, burn-in = 200, and no recombination).
Unique sequences were compared to those archived in MalAvi (Bensch et al., Reference Bensch, Hellgren and Pérez-Tris2009) and GenBank (https://www.ncbi.nlm.nih.gov/genbank/); sequences with a match lower than 100% were considered new lineages. To compare our sequences with published ones, we assembled an alignment that contained unique sequences that were 98–99% identical in MalAvi and GenBank (but restricted to hosts in the Neotropics), as well as lineages reported to infect Z. capensis. Alignment of compiled sequences was performed in Clustal X2 separately for both genera, using Leucocytozoon fringillinarum (TFUS04; GenBank: JQ815435) as outgroup.
Before estimating phylogenetic trees, we used PartitionFinder 2 (Lanfear et al., Reference Lanfear, Frandsen, Wright, Senfeld and Calcott2016) to choose the best model of nucleotide substitution for each dataset (run with branch lengths linked, all models, Akaike Information Criterion (AIC), and greedy algorithm). The best partition scheme for Plasmodium (by codon position) was TrN (Tamura-Nei) + I (invariable sites) + G (gamma distribution) (first), F81 (Felsenstein 1981) (second) and GTR (general time reversible) + G (third); for Haemoproteus, it was GTR + I + G (first), HKY (Hasegawa-Kishino-Yano) + I + G (second) and GTR + G (third). Phylogenetic analyses were performed separately by genus in MrBayes 3.2.6 (Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003) with 2 million generations, sampling each 1000 trees and burning 250, retaining 1750 trees used to obtain a 50% majority-rule consensus tree.
Morphological identification
Blood smears were examined using a Leica DM750 microscope (Leica Microsystems, Heerbrugg, Switzerland) at 400× for 10 min and then at high magnification 1000× for 20 min to detect haemoparasites. From positive slides, we obtained images representing different parasite blood stages using a Leica EC3 camera and processed with LAS EZ software (Leica Microsystems Ltd., Switzerland, 2012). Taxonomic determination of parasites was made using Valkiūnas (Reference Valkiūnas2005) and Valkiūnas and Iezhova (Reference Valkiūnas and Iezhova2018) keys as well as recent descriptions of new species (Walther et al., Reference Walther, Valkiūnas, González, Matta, Ricklefs, Cornel and Sehgal2014; Mantilla et al., Reference Mantilla, González, Lotta, Moens, Pacheco, Escalante, Valkiūnas, Moncada, Pérez-Tris and Matta2016).
Prevalence and mean parasitaemia
Prevalence was assessed based on molecular lineage identification. Values of parasitaemia (intensity of infection) were obtained from 79 blood smears of infected samples (18–20 smears randomly selected per month). Blood smears were screened under an Olympus CX31 microscope (Olympus Optical Co. Ltd., Tokyo, Japan) at 1000× magnification, counting the number of infected, uninfected and immature erythrocytes (polychromatophils) until a total of 10 000 cells per smear were examined (Waldenström et al., Reference Waldenström, Bensch, Hasselquist and Ostman2004). Prevalence and mean parasitaemia were analysed using software Quantitative Parasitology 3.0 (Rózsa et al., Reference Rózsa, Reiczigel and Majoros2000; Reiczigel and Rózsa, Reference Reiczigel and Rózsa2005). Estimation of confidence intervals (CIs) for prevalence was performed with the Sterne method (Reiczigel, Reference Reiczigel2003). Mean parasitaemia and 95% CIs were estimated with the BCa method, using 2000 bootstrap replicates (Rózsa et al., Reference Rózsa, Reiczigel and Majoros2000). Significant differences in prevalence between lineages were estimated using the unconditional exact test, whereas significant differences in mean parasitaemia between lineages were estimated using a bootstrap-t test with 2000 replicates (Rózsa et al., Reference Rózsa, Reiczigel and Majoros2000).
Factors associated with haemosporidian infections
To determine how biotic or abiotic factors are related to infection status (presence/absence of infection) and parasitaemia, and to explore the associations between infection and body condition, we performed a series of generalized linear model (GLM) analyses in R (R Development Core Team, 2008) using the MASS package (Venables and Ripley, Reference Venables and Ripley2002). Factors related to infection status were assessed using a logistic regression with binomial distribution and logit link function. Infection status was selected as dependent variable, and the following variables were selected as factors: age (adult, subadult) and body condition, as biotic factors; total monthly precipitation (mm), sampling site (S1, S2, S3, S4) and capture date, as abiotic factors. Body condition was calculated as the residuals from an ordinary least squares linear regression of body mass (dependent variable) against tarsus length (independent variable) (Green, Reference Green2001). Once the general model was obtained by including all factors, a backward AIC-based stepwise model selection was performed to find the minimal adequate model. Differences among factor levels were evaluated by examining the estimates of the model.
Factors related to parasitaemia were explored by means of a GLM assuming a negative binomial distribution to account for overdispersion of the data. Parasitaemia was selected as dependent variable and the factors were the same selected previously for analysing infection status (AIC-based model selection performed as previously). Finally, we investigated the associations among parasite lineages, coinfections and body condition. Body condition was selected as dependent variable and parasite lineage, presence of coinfections (detected by either morphology or genetic sequencing), age, date of measurement, monthly precipitation and sampling site as factors (AIC-based model selection performed as previously). In all models, multicollinearity among factors was inspected by the variance inflation factor. Goodness of fit was investigated by inspecting the difference between null and residual deviance of the models.
Results
Relative abundance and movement of individuals
We captured 871 individuals from 36 species of birds. Of this total, 226 were Z. capensis, which represented a relative abundance of 26% in mist net captures, the highest in this study. We recorded 20 recaptures, 17 within the same sampling sites, and three of birds banded at site 2 and recaptured at site 4 or vice versa.
Molecular lineage identification and phylogenetic relationships
We analysed a total of 177 samples of Z. capensis by PCR and sequencing, 135 of which were positive for Haemosporida infection (Table 1). A total of 86 samples showed single infections for Haemoproteus ZC1 (GenBank: KC480265; Jones et al., Reference Jones, Cheviron and Carling2013); 34 for Plasmodium BAEBIC02 (GenBank: KF537291; Walther et al., Reference Walther, Valkiūnas, González, Matta, Ricklefs, Cornel and Sehgal2014; González et al., Reference González, Lotta, García, Moncada and Matta2015) and four for a new lineage of Plasmodium, Plasmodium ZOCAP15 (GenBank: MK077679). Phasing of sequences showing coinfections identified five fully resolved haplotypes (no phase uncertainties and phase probabilities = 1.0); three were coinfections by Plasmodium BAEBIC02 and Haemoproteus ZC1, and two by Haemoproteus ZC1 and Plasmodium ZOCAP15. Among the four sequences that were not resolved, three were regarded as combinations of BAEBIC02 and themselves, and one was regarded as a combination of lineage ZC1 and another mixed infection in the sample, both with high uncertainty (phase probabilities > 0.51). Thus, in further analyses we only included the five, fully resolved coinfections.
Table 1. Association of molecular lineage (amplified by PCR) and morphospecies (identified by microscopy) found in Z. capensis sampled at Bosque Protector Jerusalem Quito-Ecuador. The first morphospecies denotes the morphospecies responsible for the main infection, whereas the other represents a coinfection

a Totals are different because not all samples diagnosed by PCR were diagnosed by morphology. Out of the 135 samples that were positive by PCR, four were undetermined coinfections and two were not identified to lineage.
(d/s1) number of samples detected positive for molecular lineage/number of samples screened by PCR-sequencing.
(d/s2) number of samples detected positive for morphospecies/number of samples screened by microscopy once molecular lineage was determined.
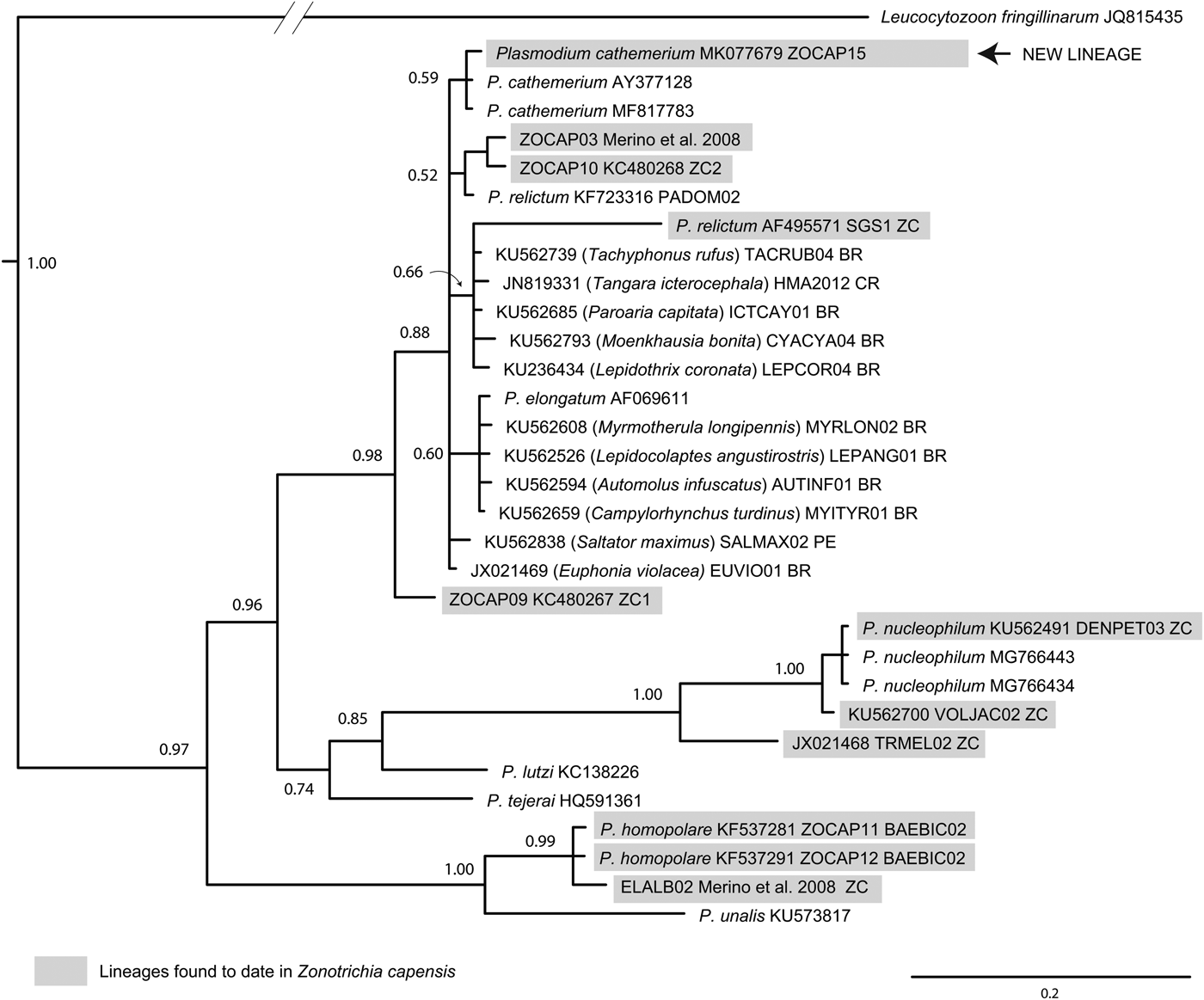
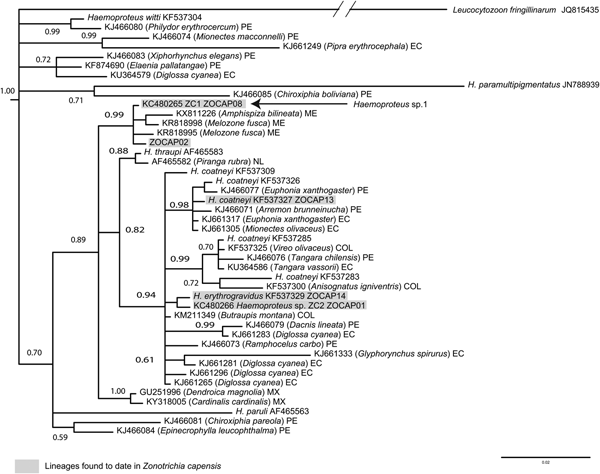
Phylogenetic analyses based on 34 unique sequences of related avian Plasmodium lineages placed the new lineage, ZOCAP15, in a clade with other P. cathemerium lineages (Fig. 2). Phylogenetic analyses based on 45 unique sequences of related avian Haemoproteus placed ZC1 as a distinctive lineage, together with lineages that infect Z. capensis and other Passeriformes, which have not been associated with any morphospecies (Fig. 3).

Fig. 2. Phylogenetic position of P. (N.) homopolare (BAEBIC02) and the new lineage, Plasmodium (H.) cathemerium (ZOCAP15), among closely related lineages of Plasmodium, based on a Bayesian phylogenetic analysis of 480 base pairs; Bayesian posterior probabilities are showed over nodes. Scientific names in parentheses are hosts were lineages have been detected.

Fig. 3. Phylogenetic position of Haemoproteus (P.) sp. 1 (ZC1) among closely related lineages of Haemoproteus, based on a Bayesian phylogenetic analysis of 480 base pairs; Bayesian posterior probabilities are showed over nodes. Scientific names in parentheses are hosts were lineages have been detected.
Morphological identification and its association with molecular identification
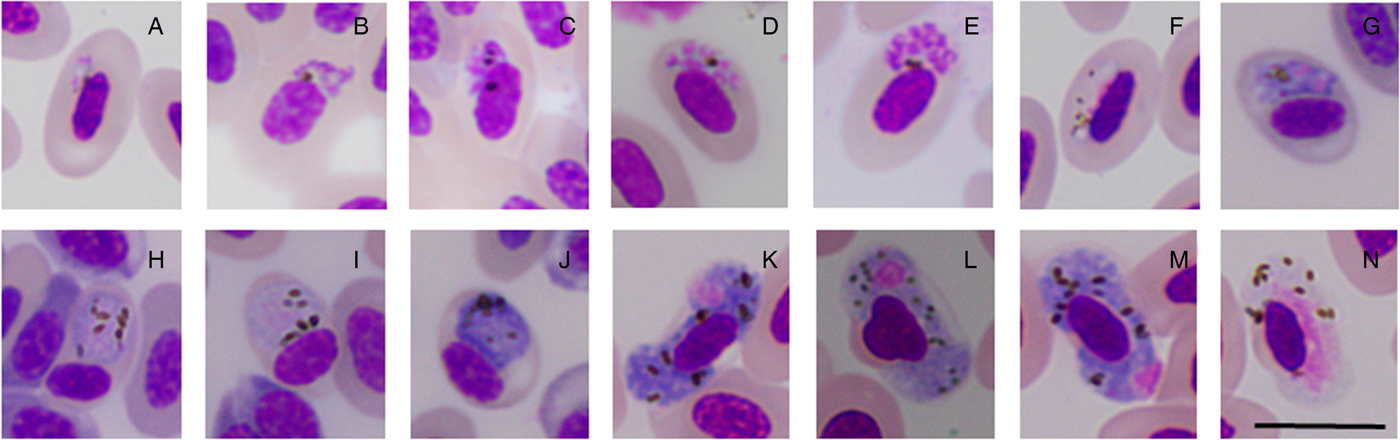
Morphological identification was performed by microscopy on a fraction of the molecularly diagnosed samples (Table 1). This analysis allowed us to identify six morphospecies infecting Z. capensis: Plasmodium (Novyella) homopolare, P. (Haemamoeba) cathemerium, P. (N.) nucleophilum, Haemoproteus (Parahaemoproteus) coatneyi, H. (P.) erythrogravidus, and an undescribed morphospecies that we call Haemoproteus (P.) sp. 1 (Fig. 4).

Fig. 4. Morphological features of some haemosporidian parasites from the blood of Zonotrichia capensis. Plasmodium (N.) nucleophilum: (A) trophozoites, (B–E) erythrocytic meronts, (F) young microgametocytes. Plasmodium (H.) cathemerium: (G) erythrocytic meront, (H, I) microgametocytes, (J) macrogametocyte. Haemoproteus (P.) sp. 1: (K–M) macrogametocytes, (N) microgametocyte. Giemsa-stained blood films. Scale bar = 10 µm.
From 74 screened samples with single infections for molecular linage Haemoproteus (P.) ZC1, 64 showed single microscopic infections by Haemoproteus (P.) sp. 1 (Table 1); thus, hereafter, we refer to this molecular lineage as Haemoproteus (P.) sp1 (ZC1). Out of 25 screened samples with single infections by molecular linage Plasmodium BAEBIC02, 20 samples were only infected by morphospecies P. (N.) homopolare (Table 1); thus, hereafter, we refer to the molecular lineage as P. (N) homopolare (BAEBIC02). One sample infected with lineage ZOCAP15 was heavily infected by Plasmodium (H.) cathemerium and H. (P.) erythrogravidus (Table 1). Given that phylogenetic analyses placed the new lineage, ZOCAP15, in a clade with other P. (H.) cathemerium lineages and the screened sample was infected with Plasmodium (H.) cathemerium, hereafter, we refer to this molecular lineage as P. (H.) cathemerium (ZOCAP15). Nine samples with molecular coinfections showed single infections by microscopy, whereas 16 samples with molecular single infections showed coinfections by microscopy (Table 1).
Prevalence and mean parasitaemia
Prevalence of haemosporidian parasites in Z. capensis was 76.3% based on molecular diagnosis (Table 2). Prevalence by lineage considered samples bearing single infections or fully resolved coinfections. Prevalence for Haemoproteus (P.) sp. 1 (ZC1) was significantly higher than for P. (N.) homopolare (BAEBIC02) (exact P value [two-tailed] < 0.0001). Mean parasitaemia for avian haemosporidians was 84.1 infected erythrocytes/10 000 erythrocytes counted (Table 2), whereas mean parasitaemia was also significantly higher for Haemoproteus (P.) sp. 1 (ZC1) than for P. (N.) homopolare (t-statistic = −2.789; bootstrap P value [two-tailed] = 0.011).
Table 2. Prevalence and parasitaemia by haemosporidian parasites in Z. capensis

a Prevalence of infection was calculated based on molecular results and includes single infections and unambiguous coinfections by molecular lineage.
b Parasitaemia (infected erythrocytes in 10 000 cells counted) was calculated based on single infections by lineage and morphospecies; total parasitaemia (all lineages) includes individuals with single and mixed infections from any lineage or morphospecies. n: number of samples for which parasitaemia was analysed by microscopy; –: not calculated because of low sampling size.
Factors associated with haemosporidian infections
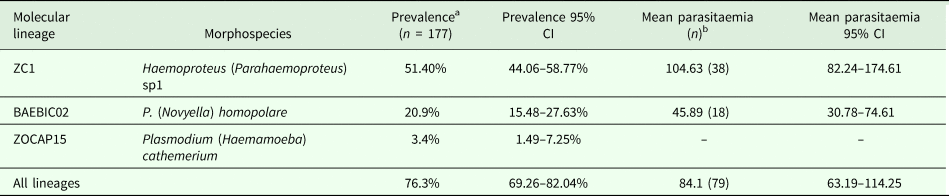
When analysing the predictors of infection status, AIC-based model selection resulted in a model that included total monthly precipitation, age and site as predictors. Model estimates indicated that more individuals were infected on months with higher precipitation (estimate = 0.01, z-value = 2.25, P < 0.02); more adults than subadults were infected (estimate = 2.18, z-value = 4.25, P < 0.0001); and there were less individuals infected on site 2 compared to individuals in sites 1 and 4 (estimate = −1.51, z-value = −2.01, P = 0.04) but not from site 3 (Fig. 5A). The model explained 13.3% of the variation in infection status.

Fig. 5. Effect plots from models investigating infection status (A), intensity of infection (B) and body condition (C). Grey shaded area and the error bars indicate standard error (s.e.).
For the analysis of parasitaemia, a global model was fitted by assuming a Poisson distribution; however, the analysis of the residual deviance and the degrees of freedom detected overdispersion. Thus, to correct for overdispersion we applied a negative binomial distribution. AIC-based stepwise model selection identified a final model that only retained site as factor. Here, our results indicated that individuals from site 2 presented the lowest parasitaemia (estimate = −1.65, z-value = −3.11, P < 0.0001) (Fig. 5B). The model explained 11.1% of the variation in parasitaemia.
In the analysis of associations of parasite lineages and coinfections with body condition, stepwise AIC-based model selection resulted in a model where the presence of coinfections and site were selected as factors (no relationship between parasite lineage and body condition was observed). Estimates from the minimal adequate model showed individuals from sites 2 and 4 (site 2: estimate = 1.19, t-value = 3.02, P < 0.01, site 4: estimate = 1.33, t-value = 3.26, P < 0.01), and individuals with presence of coinfections (estimate = 0.65, t-value = 1.97, P = 0.05) as having better body condition (Fig. 5C). The model explained 14.8% of the variation in body condition. No multicollinearity was observed among factors in all models.
Discussion
Diversity of haemosporidians
We identified three molecular lineages of haemosporidian parasites: Plasmodium (N.) homopolare (BAEBIC02), Haemoproteus (P.) sp1 (ZC1) and Plasmodium (H.) cathemerium (ZOCAP15). Lineage Plasmodium (N.) homopolare (BAEBIC02) was first isolated from Baeolophus bicolor (Paridae) in the United States (Martinsen et al., Reference Martinsen, Sidor, Flint, Cooley and Pokras2007) and it has been found infecting Z. capensis in Peru (Marzal et al., Reference Marzal, García-Longoria, Cárdenas Callirgos and Sehgal2015) and Colombia (Walther et al., Reference Walther, Valkiūnas, González, Matta, Ricklefs, Cornel and Sehgal2014; González et al., Reference González, Lotta, García, Moncada and Matta2015), as well as other 14 species of passerines from Alaska to Peru (Galen and Witt, Reference Galen and Witt2014; Oakgrove et al., Reference Oakgrove, Harrigan, Loiseau, Giers, Seppi and Sehgal2014; Walther et al., Reference Walther, Valkiūnas, González, Matta, Ricklefs, Cornel and Sehgal2014; González et al., Reference González, Lotta, García, Moncada and Matta2015). Haemoproteus sp. ZC1 infects Z. capensis in Peru (Jones et al., Reference Jones, Cheviron and Carling2013), other Emberizidae in México (Reinoso-Pérez et al., Reference Reinoso-Pérez, Canales-Delgadillo, Chapa-Vargas and Riego-Ruiz2016; Ham-Dueñas et al., Reference Ham-Dueñas, Chapa-Vargas, Stracey and Huber-Sannwald2017), and Troglodytes aedon in Peru (Galen and Witt, Reference Galen and Witt2014). This lineage shows considerable genetic distance from those of other Haemoproteus morphospecies (Fig. 3), making the associated morphospecies a good candidate for morphological description.
The finding of the new lineage, ZOCAP15, couples with the first report of the Plasmodium (H.) cathemerium morphospecies in Z. capensis. Its low prevalence may indicate that it is either rare in this population or highly virulent. One individual carrying this parasite was analysed by microscopy and showed one of the highest parasitaemias our study (481 infected cells/10 000 cells counted). Plasmodium (H.) cathemerium Hartman, 1927 is a generalist parasite with a wide range of hosts (Bennett et al., Reference Bennett, Bishop and Peirce1993; Valkiūnas, Reference Valkiūnas2005) that may cause severe forms of malaria (Gabaldón et al., Reference Gabaldón, Ulloa and Zerpa1988; Valkiūnas, Reference Valkiūnas2005; Vanstreels et al., Reference Vanstreels, da Silva-Filho, Kolesnikovas, Bhering, Ruoppolo, Epiphanio, Amaku, Junior and Martins Braga2015). Thus, it would be important to investigate the prevalence of Plasmodium (H.) cathemerium in this community, not only in Z. capensis – which is definitively a competent host, as gametocytes were observed in the blood smear – but also in other species.
Together with previous studies (González et al., Reference González, Lotta, García, Moncada and Matta2015; Mantilla et al., Reference Mantilla, González, Lotta, Moens, Pacheco, Escalante, Valkiūnas, Moncada, Pérez-Tris and Matta2016), our results confirm at least six morphospecies of haemosporidians infecting Z. capensis and add to the current knowledge by associating morphospecies Haemoproteus (P.) sp1 to molecular lineage ZC1. Interestingly, three morphospecies not detected by molecular diagnosis – Plasmodium (N.) nucleophilum, H. (P.) coatneyi and H. (P.) erythrogravidus – were detected by microscopy in coinfection with P. (N.) homopolare or Haemoproteus (P.) sp1. This result may be caused by differences in parasitaemia or by the differential ability of the primers to amplify parasite DNA in mixed infections, especially when both Haemoproteus and Plasmodium are present (Bernotienė et al., Reference Bernotienė, Palinauskas, Iezhova, Murauskaitė and Valkiūnas2016).
Given that our sampling effort for a single locality has been considerably higher than in previous studies of Z. capensis (e.g. Jones et al., Reference Jones, Cheviron and Carling2013; González et al., Reference González, Lotta, García, Moncada and Matta2015; Marzal et al., Reference Marzal, García-Longoria, Cárdenas Callirgos and Sehgal2015), our results may be reflecting the actual diversity of haemosporidians in this population. Rather than the low diversity initially expected, we found the highest diversity of haemosporidian parasites infecting Z. capensis on a single site. Additionally, coinfections were relatively frequent compared to previous studies (e.g. Cosgrove et al., Reference Cosgrove, Wood, Day and Sheldon2008; Levin et al., Reference Levin, Zwiers, Deem, Geest, Higashiguchi, Iezhova, Jimenez-Uzcategui, Kim, Morton, Perlut, Renfrew, Sari, Valkiūnas and Parker2013; Ferraguti et al., Reference Ferraguti, Martínez-de la Puente, Bensch, Roiz, Ruiz, Viana, Soriguer and Figuerola2017; but see Marzal et al., Reference Marzal, Bensch, Reviriego, Balbontin and de Lope2008). This condition was detected in 7% of molecular diagnoses and 15% of microscopic diagnoses (Table 1), stressing the need for both kinds of diagnoses in the survey of haemoparasites (see Carlson et al., Reference Carlson, Nelms, Barker, Reisen, Sehgal and Cornel2018; Pacheco et al., Reference Pacheco, Matta, Valkiunas, Parker, Mello, Stanley, Lentino, Garcia-Amado, Cranfield, Pond and Escalante2018).
Prevalence and parasitaemia
Prevalence of haemosporidians in Z. capensis in this Andean dry forest (76%) is higher than that reported by Jones et al. (Reference Jones, Cheviron and Carling2013) in Andean Peru (42%) and similar to that found by Escallón et al. (Reference Escallón, Weinstein, Tallant, Wojtenek, Rodríguez-Saltos, Bonaccorso and Moore2016) in the Andean foothills of the Amazon (73%). Prevalence of lineage P. (N.) homopolare (BAEBICO2) (19.2%) is higher than that reported by Marzal et al. (Reference Marzal, García-Longoria, Cárdenas Callirgos and Sehgal2015) for Z. capensis in the eastern slope of the Andes of Peru (15.15%), whereas prevalence of Haemoproteus sp1 (ZC1) (48.6%) was also higher than that reported by Jones et al. (Reference Jones, Cheviron and Carling2013) in Peru (25%). Overall, high prevalence of haemosporidians in our study could be related to habitat, latitude, regional or community-level assemble of parasites (e.g. Fecchio et al., Reference Fecchio, Svensson-Coelho, Bell, Ellis, Medeiros, Trisos, Blake, Loiselle, Tobias, Fanti, Coffey, de Faria, Pinho, Felix, Braga, Anciaes, Tkach, Bates, Witt, Weckstein, Ricklefts and Farias2017), or high abundance of Z. capensis, the highest in the avian community of the study site (see Ellis et al., Reference Ellis, Medeiros, Collins, Sari, Coffey, Dickerson, Lugarini, Stratford, Henry, Merrill, Matthews, Hanson, Roberts, Joyce, Kunkel and Ricklefts2017). Higher prevalence and parasitaemia of Haemoproteus sp1 (ZC1) over P. (N.) homopolare (BAEBICO2) might be related to the relatively mild or sublethal effects previously reported for Haemoproteus (Garvin et al., Reference Garvin, Homer and Greiner2003), but further studies are necessary to confirm the pathology of this lineage infecting Z. capensis.
Factors associated with haemosporidian infections
Our analyses produced both expected and puzzling results. Infection status was predicted by monthly precipitation, age and sampling site. First, a positive effect of precipitation on infection status is likely to be related to better conditions for vector reproduction, which would increase the acquisition of new infections or trigger higher parasitaemia within an already infected host (Cornet et al., Reference Cornet, Nicot, Rivero and Gandon2014). Although higher prevalence has been tied to reproduction of the host (e.g. Knowles et al., Reference Knowles, Wood, Alves, Wilkin, Bensch and Sheldon2011; Ham-Dueñas et al., Reference Ham-Dueñas, Chapa-Vargas, Stracey and Huber-Sannwald2017), this scenario is not plausible given the apparently continuous reproduction of Z. capensis at the study site (Cadena-Ortiz, personal observation), which contrasts with reports of reproduction seasonality in two other nearby localities, but at higher elevation (Papallacta at 3300 m a.s.l. and Pintag at 2900 m a.s.l.; Moore et al., Reference Moore, Bonier and Wingfield2005). Future studies should aim at monitoring reproductive activities of the host, prevalence and parasitaemia, as well as abundance and diversity of vectors in this locality, simultaneously. Second, we found more infections among adults than among subadults, which has been found in previous studies (e.g. Wood et al., Reference Wood, Cosgrove, Wilkin, Knowles, Day and Sheldon2007). This difference is usually attributed to the increased probability of acquiring and maintaining chronic infections with age or, alternatively, higher mortality of subadults (Davidar and Morton, Reference Davidar and Morton1993). Third, we found that fewer individuals were infected on site 2. This result agrees with the lower parasitaemia found on site 2. Lower prevalence and parasitaemia on site 2 could result from lower abundance of vectors within the Citrus sp.—P. americana plantation. In fact, habitats with more natural vegetation may favour higher richness and abundance of vector species (Culicoides and Cuilicidae) through the availability of breeding places (Mullen, Reference Mullen, Mullen and Durden2002; Eldridge, Reference Eldridge and Marquardt2005). Thus, areas with less natural vegetation, like site 2, could have lower abundance and diversity of vectors, explaining lower prevalence and parasitaemia on this site.
When exploring the predictors of body condition, we found that individuals bearing coinfections, and those from sites 2 and 4, had better body condition. Recaptures in the study area reflect that there is only limited movement of Z. capensis among sites, explaining why sites seem to be acting as independent ‘islands’, with a few exceptions of movement between sites 2 and 4 (which are relatively closer). Given that individuals from site 2 had less prevalence and parasitaemia, it is not surprising that they have better body condition (although body condition could not predict levels of prevalence and parasitaemia); however, we may not explain why individuals from site 4 had better body condition. It is also difficult to explain why individuals with coinfections would show better body condition than those with single infections (although results are marginally significant). This is specially puzzling since mixed infections are expected to have more detrimental effects on body condition because of higher compromise of the immune system (Marzal et al., Reference Marzal, Bensch, Reviriego, Balbontin and de Lope2008).
Concluding remarks
The low percentages of variation explained by our models suggest that variables not measured in this study may affect infection status, parasitaemia and body condition in the rufous-collared sparrow. Still, our results support the idea that Z. capensis could be a good model organism for studying the dynamics of avian haemosporidian infections given its high abundance, apparently high resilience to infections, and relatively high diversity of parasites (and coinfections). Future studies should analyse a more comprehensive list of variables and interactions, as well as a greater sample, in order to contribute to a more integrative understanding of avian haemosporidian infections in this host.
Acknowledgements
Jefferson García and Judit Mateos were instrumental in obtaining field data, and Alma Hernández, Cristian Andrade, Cristian Poveda, Cristina Toapanta, Daniel Arias, Daniel Rivadeneira, Gabriela Galarza, Ibeth Alarcón, Josue Arteaga, María Galbarro, Marta Quitian and Viviana Jaramillo, helped also during fieldwork. Previous versions of this manuscript benefited from comments by Tjitte de Vries. Ángel Orellana and Danilo Armas and personnel of Bosque Protector Jerusalem, provided access to the field area and logistic support. Nicolás Peñafiel performed final molecular analyses to discard false negatives. Javier Pérez-Tris, Michaël Möens, Matthew Carling, Rafael Narváez, Anahi Paca, Gabriela Lugo, Gabriela Toscano, Javier Fajardo and Gorki Ríos provided insights into field procedures, data management, maps or analyses. Analysis of genetic samples was authorized by Ministerio del Ambiente of Ecuador, through Contrato de Acceso a Recursos Genéticos (MAE-DNB-CM-2015-0017) granted to Universidad Tecnológica Indoamérica.
Financial support
This study was supported by Universidad Tecnológica Indoamérica (Convocatorias a Proyectos 2012, 2014) and Universidad San Francisco de Quito (HUBI 12434).
Conflict of interest
None.
Ethical standards
Bird manipulation performed during this study followed both ethical and legal procedures, and tried to minimize suffering and stress.
Author ORCIDs
Héctor Cadena-Ortiz 0000-0003-4653-2072; Nubia E. Matta 0000-0003-1775-0804; Elisa Bonaccorso 0000-0002-7262-9356