




I. INTRODUCTION
Throughout the past ten years, a great deal of condensed aminomercaptotriazole derivatives with analgesic/anti-inflammatory and cytotoxic activity have been synthesized (Aytaç et al., Reference Aytaç, Tozkoparan, Kaynak, Aktay, Göktaş and Ünüvar2009, Reference Aytaç, Durmaz, Houston, Çetin-Atalay and Tozkoparan2016; Tozkoparan et al., Reference Tozkoparan, Aytaç and Aktay2009, Reference Tozkoparan, Aytac, Gursoy, Gunal and Aktay2012). In a previous work (Aytaç et al., Reference Aytaç, Durmaz, Houston, Çetin-Atalay and Tozkoparan2016) reported that a series of triazolothiadiazines, display significant in vitro cytotoxic activity on human epithelial cancers, especially liver cancer. In order to add new members to this promising chemical series, new analog compounds, named 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole, have been prepared by bioisosteric replacement of the triazolothiadiazine ring with a triazolothiadiazole one. The compound is one example of this series having a cytotoxic activity which is obtained as powder. The crystal structure determination of the compound was accomplished by X-ray powder diffraction (PXRD).
We introduce here, the preparation and structural characterization of 3-[1-[4-(2-methylpropyl)phenyl]ethyl]-6-(4-fluorophenyl)-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole, as well as structure solution by laboratory X-ray powder diffraction data (PXRD) supported by the density functional theory (DFT) calculations. We performed a comprehensive first principles study on the structural properties of the compound. Structure optimization calculations were completed with both pure DFT and dispersion-corrected density functional theory (DFT-D). It is important to consider the van der Waals effects for molecular crystal structures, therefore optB88-vdW, B3LYP-D2, and PBE-D2 functionals were realized. Finally, the geometric parameters of the crystal structure obtained by Rietveld refinement were compared with the optimized structures.
II. EXPERIMENTAL
A. Instrumentation
Melting heat was determined on a Thomas Hoover capillary melting point apparatus (Philadelphia, PA, USA) and was uncorrected. IR spectra were recorded on a Perkin Elmer 1720X FT-IR spectrometer (Beaconsfield, UK) by direct sampling of the compounds. 1H-NMR spectra were recorded on a Varian Mercury 400, a 400-MHz High-Performance Digital FT-NMR instrument (Varian, Palo Alto, CA, USA) in CDCl3 using tetramethylsilane as an internal standard. High-Resolution Mass Spectra (HRMS) data were collected with a Waters Micromass ZQ LC-MS Spectrometer (Milford, MA, USA) instrument using the ESI (+) method. Elementary analysis of the resulting compound was performed with Leco CHNS 932 analyzer at Ankara University, Faculty of Pharmacy Central II Laboratory and data were determined within ±0.4% of the theoretical values.
B. Synthesis of the compounds
The preparation of the title compound was carried out in two steps (Figure 1). Firstly, commercially available ibuprofen reacted with thiocarbohydrazide at its melting point (about 170 °C) to obtain aminomercaptoriazole derivative (Aytaç et al., Reference Aytaç, Durmaz, Houston, Çetin-Atalay and Tozkoparan2016). In the second step, the reaction of aminomercaptoriazole scaffold with 4-fluoro benzoic acid in phosphorus oxychloride via a one-pot two-component reaction gave target compound (Tozkoparan et al., Reference Tozkoparan, Aytac, Gursoy, Gunal and Aktay2012). The structure of the new compound was fully established by IR, 1H-NMR, mass spectrometry, and elemental analysis. All data of the compound were in full agreement with the proposed structure.

Figure 1. The synthesis scheme of the compound.
3-[1-[4-(2-Methylpropyl)phenyl]ethyl]-6-(4-fluorophenyl)-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole
0.20 g (52.57%). M.p.: 151–152 °C. IR: νmax: 1601 (C=N), 1308 (C–N) cm−1; 1H-NMR (CDCl3) δ (ppm) 0.87 (6H, d, (CH 3)2–CH–), 1.80–1.84 (1H, m, (CH3)2–CH–CH2–), 1.91 (3H, d, CH 3–CH–), 2.43 (2H, d, –CH–CH 2–), 4,65 (1H, q, CH3–CH–), 7.10 (2H, d, arom. H), 7.20 (2H, t, arom. H), 7.34 (2H, d, arom. H), 7.79–7.83 (2H, m, arom. H); MS (ESI+) m/z Calcd. for C21H22FN4S (M+H)+ 381.15, found 381.13; Anal. Calcd. for C21H21FN4S: C, 66.29; H, 5.56; N, 14.73; S, 8.43. Found: C, 66.57; H, 5.72; N, 14.86; S, 8.57.
C. PXRD data collection
PXRD of the compound was collected in Debye–Scherrer type geometry using Bruker D8 Advance powder diffractometer operated at 48 kV, 38 mA with molybdenum radiation (λ Kα1 = 0.709 Å, λ Kα2 = 0.71 Å) and equipped with a Göbel-mirror on the primary side monochromator and a position sensitive silicon-strip detector named “Lynx-Eye”. The following optics were used: primary beam soller slit (0.4 mm), divergence slit (0.4 mm), and receiving soller slit (1.5°). The powder sample of the compound loaded in a 0.7-mm diameter glass capillary after griding using a mortar. The diffracted intensities were collected at room temperature (295.0 K) in the range from 0° to 40° (2θ) in step-scan mode with a step size of 0.008° and a counting time of 15 s/step.
D. PXRD data analysis
Indexing of the PXRD pattern was carried out using the TOPAS Academic program v5.1 (Coelho, Reference Coelho2012), yielding the following values for triclinic space group 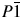 $P\bar{1}\;$ of the compound: a = 5.576 Å, b = 9.281 Å, c = 19.486 Å, α = 99.33°, β = 91.95°, γ = 98.18°, V = 983.30 Å3, and GOF = 15.14.
$P\bar{1}\;$ of the compound: a = 5.576 Å, b = 9.281 Å, c = 19.486 Å, α = 99.33°, β = 91.95°, γ = 98.18°, V = 983.30 Å3, and GOF = 15.14.
Then, Pawley refinement for the whole powder pattern of the compound was performed to confirm the choice of space group as well as the unit cell parameters. The unit cell parameters, R-factors and goodness-of-fit indicator were obtained as a = 5.5866(6) Å, b = 9.300(1) Å, c = 19.470(7) Å, α = 99.259(15)°, β = 92.001(15)°, γ = 98.159(11)° and V = 986.6(4) Å3, R wp = 3.034%, R exp = 1.078% and χ 2 = 2.814 with a good visual fit of the whole pattern. PXRD of the compound is given in Supplementary Table S1. The d obs values in the table are taken directly from the PXRD. The peak that has the strongest intensity in the entire pattern is assigned of 100 and other lines are scaled relative to this value for the data set.
A template of the molecule was built and full geometry optimization at DFT/B3LYP level using the 6–31G++(d,p) basis set was performed with Gaussian 09 program (Frisch et al., Reference Frisch, Trucks, Schlegel, Scuseria, Robb, Cheeseman, Scalmani, Barone, Mennucci, Petersson, Nakatsuji, Caricato, Li, Hratchian, Izmaylov, Bloino, Zheng, Sonnenberg, Hada, Ehara, Toyota, Fukuda, Hasegawa, Ishida, Nakajima, Honda, Kitao, Nakai, Vreven, Montgomery, Peralta, Ogliaro, Bearpark, Heyd, Brothers, Kudin, Staroverov, Kobayashi, Normand, Raghavachari, Rendell, Burant, Iyengar, Tomasi, Cossi, Rega, Millam, Klene, Knox and Cross2009). The molecule without hydrogen atoms was used as a fragment to solve the initial structure with the Monte Carlo simulated annealing (parallel tempering algorithm) method implemented in FOX program (Favre-Nicolin and Černý, Reference Favre-Nicolin and Černý2002). During the simulated annealing calculations, global parameters defining the orientation and the position of each fragment are varied during the procedure, together with torsion angles. The best solution with 62 788.61 overall best cost was obtained.
Having obtained the initial structure and profile terms, the Rietveld refinement in 2θ range from 1° to 35° was carried out using TOPAS Academic program v5.1. The background was fitted using 5-order Chebyshev polynomials. The peak asymmetry was fitted by the simple axial divergence model of Cheary and Coelho (Reference Cheary and Coelho1998). The peak profiles modeled by the Double-Voigt approach (Balzar, Reference Balzar1993; Coelho, Reference Coelho2012). During the refinement, unit-cell parameters, background, zero-point error, scale factor, peak width, and asymmetry parameters were refined together with non-H atomic coordinates. Soft restraints to all non-H atom bond lengths and angles, as well as soft planar group restraints to all rings, were applied. The values used in the soft restraints on the bond lengths and angles were taken from the optimized structure with local density approximation (LDA) functional (van de Streek, Reference van de Streek2015).
Afterwards, the coordinates of the S and F atoms were refined one by one. The atoms of each ring were refined together followed by one ring after another. For all non-H atoms, an overall isotropic atomic displacement parameter (B iso) was determined and refined. Before the final refinement, the hydrogen atoms were introduced from geometrical arguments (C–H = 0.96 Å and B iso (H) = 1.2 B iso (C)) by using Crystals program (Cooper et al., Reference Cooper, Thompson and Watkin2010). The fractional coordinates of the hydrogen atoms were refined by riding motion approximation with a macro implemented in TOPAS. Their isotropic atomic displacements (B iso) were constrained to be 1.2 times the value of the carrier atom. The Rietveld refinement was converged with a smooth difference curve (Figure 2). The structure refinement details are summarized in Table I.

Figure 2. Plot of the final Rietveld refinement of the compound. Black crosses represent the observed data, the red line indicates the calculated pattern, and the blue line at the bottom represents the difference between the observed and calculated patterns. Green vertical bars indicate Bragg reflections.
TABLE I. Crystal data and structure refinement parameters of the compound.

E. Computational methods
The correctness of the experimental crystal structures has been validated previously for 241 organic compounds, which shows that DFT calculations are an effective tool for the reproduction of molecular structures (van de Streek and Neumann, Reference van de Streek and Neumann2010). Due to the relatively low amount of powder XRD data (with an upper 2θ limit of 35°), it was aimed to perform DFT calculations in order to complement and verify the experimental structure solution of the compound.
In the theoretical calculations, the crystal structure was modeled using the first principles calculations based on DFT as performed in Vienna ab initio simulation package (VASP) (Kresse and Hafner, Reference Kresse and Hafner1993; Kresse and Furthmüller, Reference Kresse and Furthmüller1996a, Reference Kresse and Furthmüller1996b). Starting from the experimental structure solution, the crystal structure optimizations were carried out with fixed unit cell parameters. The electron–ion interactions were described by projector augmented waves (PAW) (Blöchl, Reference Blöchl1994; Kresse and Joubert, Reference Kresse and Joubert1999) with plane-waves up to the energy of 800 eV. The effects of exchange-correlations were examined by using the LDA (CA-PZ) (Ceperley and Alder, Reference Ceperley and Alder1980; Perdew and Zunger, Reference Perdew and Zunger1981) and the generalized gradient approximation (GGA) in the parametrization of Perdew–Burke–Ernzerhof (PBE) (Perdew et al., Reference Perdew, Burke and Ernzerhof1996) functional. In addition to these functionals, the van der Waals (vdW) functional (optB88-vdW) (Klimeš et al., Reference Klimeš, Bowler and Michaelides2010) was also utilized. By using the optB88-vdW functional, the PBE functional was modified to generate the PAW core potentials. Additionally, DFT-D2 developed by Grimme (Reference Grimme2006) was also used. Brillouin zone is sampled with 5 × 3 × 1 Monkhorst–Pack (Monkhorst and Pack, Reference Monkhorst and Pack1976). The relaxations of all structures were performed by using the conjugate gradient method. The total energies of all atoms in the system were converged to 10−6 eV atom−1. The energy values of the optimized structures of the compound were given in Table II.
TABLE II. Energy values of the optimized structures of the compound.

III. RESULTS AND DISCUSSION
The Rietveld refined and the DFT-optimized structures are overlaid for the comparison shown in Figure 3. The root-mean-square (r.m.s.) differences between the coordinates of the Rietveld refined and the DFT-optimized structures for the non-H atoms are 0.219 Å with LDA, 0.181 Å with PBE, 0.188 Å with B3LYP-D2, 0.189 Å with PBE-D2, and 0.177 Å with optB88-vdW functional, which are in the range expected for correct powder structures obtained from the laboratory PXRD [Note: It should be below 0.25 Å (van de Streek and Neumann, Reference van de Streek and Neumann2014)]. The conservation of the packing arrangement and the good agreement between the refined and the optimized structures of the compound supports the validity of our structure solution (van de Streek and Neumann, Reference van de Streek and Neumann2014).

Figure 3. Comparison of the experimental crystal structure from powder data (red) and the calculated structure by DFT investigations with LDA (orange), optB88-vdW (brown), PBE (pink), B3LYP-D2 (blue), and PBE-D2 (green) functionals projected onto ac plane.
Long-range dispersive interactions (van der Waals interactions) are particularly important for prediction of the organic crystal structures. Even though the pure DFT calculations could give good results, it is limited to describe correctly van der Waals interactions. Since an accurate description of dispersion interactions provide an advantage, DFT calculations with van der Waals corrections and functional were performed. In the literature, it is not observed previously that the structure solution obtained from laboratory PXRD was supported by the optB88-vdW functional. A large number of experimental organic crystal structures were verified by using the DFT-D method (van de Streek and Neumann, Reference van de Streek and Neumann2010). In our study, the experimental structure was compared with the optimized structures obtained with the pure DFT, DFT-D2, and optB88-vdW functionals. The best match was gained from the optimization with optB88-vdW functional.
The bond lengths and angles within the triazolo-thiadiazole system of the refined structure agreed well with the reference structures (Lu et al., Reference Lu, Zhang, Song, Tan and Shao2008; Khan et al., Reference Khan, Hameed, Tahir, Bokhari and Khan2009, Reference Khan, Zaib, Ibrar, Rama, Simpson and Iqbal2014; Fan et al., Reference Fan, Yang, Zhang, Mi, Wang, Cai, Zuo, Zheng and Song2010; Cansız et al., Reference Cansız, Cetin, Orek, Karatepe, Sarac, Kus and Koparir2012; Wu, Reference Wu2013; Gündoğdu et al., Reference Gündoğdu, Aytaç, Müller, Tozkoparan and Kaynak2017) and the optimized structures (Table III). The Ortep-3 (Farrugia, Reference Farrugia2012) diagram of the compound with thermal ellipsoids at the 50% probability level is shown in Figure 4.

Figure 4. View of the title molecule showing the atomic numbering and 50% probability displacement spheres.
TABLE III. Selected bond lengths and angles (Å, °) for the experimental (PXRD) and optimized structures of the compound.

In the discussion part of the geometry, the DFT optimized structure with optB88-vdW functional was used, because the hydrogen bonds (Table IV) are analyzed better in the optimized structures (Kaduk et al., Reference Kaduk, Gindhart and Blanton2016). As expected, the 1,2,4-triazolo, 1,3,4-thiadiazole, and phenyl rings are essentially planar. The highest deviations from the planarity are 0.001 Å, 0.004 Å, 0.005 Å, and 0.008 Å, respectively for the 1,2,4-triazolo ring, the 1,3,4-thiadiazole ring, the C16–C21 ring, and the C5–C10 ring. An intermolecular C17–H171⋯S1 interaction supports the planarity of the molecular structure.
TABLE IV. Hydrogen bonding geometry (Å, °) for the experimental (PXRD) and optimized structures of the compound.

Symmetry codes: (i) x − 1, y, z; (ii) −x + 1, −y + 1, −z + 1.
The 1,2,4-triazolo ring is almost coplanar with the 1,3,4-thiadiazole ring with a mean deviation of 0.005 Å, which forms a dihedral angle of 0.53°. The dihedral angles between the triazole-thiadiazole ring system and the planes of the C16–C2 and the C5–C10 rings are 19.72° and 66.36°, respectively.
There are two C–H⋯π intermolecular hydrogen-bonding interactions. The first of them is between the atom H123 and the centroid of the 1,3,4-thiadiazole ring with a distance of 2.71 Å (symmetry code: 1 − x,1 − y,1 − z) and the centroid angle is 129° (Supplementary Figure S1). The second one is between the atom H171 and the centroid of the C5–C10 ring and the distance is 2.80 Å (symmetry code: 1 − x, 2 – y, 1 – z) and the centroid angle is 140° (Supplementary Figure S2). There is a chiral center at C11 atom in the structure.
The first short intermolecular contact of the triazolothiadiazole ring containing structures in the literature was observed in the study of Khan et al. (Reference Khan, Bakht, Ibrar, Abbas, Hameed, White, Rana, Zaib, Shahid and Iqbal2015) as 2.795(2) Å, which is shorter than the van der Waals radii sum of nitrogen and sulfur atoms (3.35 Å) for S⋯N. The short intermolecular contact is also found in the title structure (between the symmetry codes: x, y, z and 2 – x, 2 – y, 1 – z) for S1⋯N2 with 2.96 Å.
In addition, weak π–π stacking interactions between the 1,3,4-thiadiazole ring and 1,2,4-triazolo ring (between symmetry codes: x, y, z and 1 – x, 2 – y, 1 – z) with a centroid–centroid distance of 3.4345 Å is observed. The two C–Hπ interactions, short intermolecular contacts (S⋯N and C⋯N), π–π stacking interactions and inter molecular hydrogen bonds (Table IV) link the molecules into a three-dimensional network (Supplementary Figure S3).
IV. CONCLUSION
The preparation of the originally synthesized compound, structural characterization and its crystal structure achieved from the PXRD are reported in this study, which is consistent with the DFT calculations. When compared both pure DFT and DFT-D results, the best agreement with the refined structure was achieved by optB88-vdW functional. To the best of our knowledge, there is no study before by validation with optB88-vdW functional of the experimental structure solution obtained from the PXRD. The good agreement between the refined and optimized structures (Figure 3) is the strong evidence that the structure is correct.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0885715619000654.
ACKNOWLEDGMENTS
Financial support to Gülsüm Gündoğdu from The Scientific and Technological Research Council of Turkey (TÜBİTAK) is gratefully acknowledged (2214-A one-year international research grant during PhD). She is indebted to Professor Hermann Gies and his work group for Crystal Chemistry from the Ruhr University Bochum, Institute for Geology, Mineralogy and Geophysics, Department of Crystallography, for hosting her during the period of one year. The authors thank also to Hacettepe University Research Center to purchase the TOPAS Academic program (Project ID: 9460, FHD-2016-9460). The numerical calculations reported in this paper were fully performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources).










