INTRODUCTION
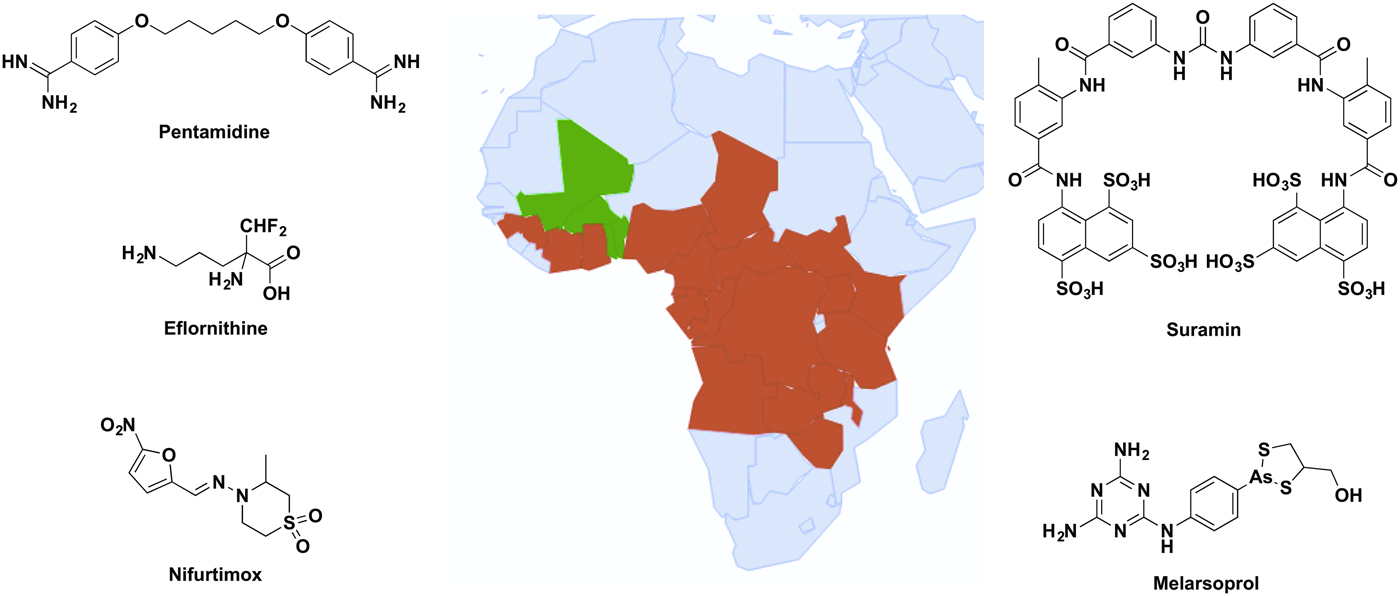
Up to 70 million people in sub-Saharan Africa are at risk of contracting human African trypanosomiasis (HAT) (Simarro et al. Reference Simarro, Cecchi, Franco, Paone, Diarra, Ruiz-Postigo, Fèvre, Mattioli and Jannin2012), also known as African sleeping sickness, caused by the kinetoplastid parasite Trypanosoma brucei. Two subspecies of the parasite cause disease in humans; T. brucei gambiense in West Africa and T. brucei rhodesiense in East Africa, both of which are spread by the tsetse fly. Both forms are fatal if untreated and are estimated to cause up to 20 000 cases of HAT per year (World Health Organization, 2013). Trypanosoma brucei evades the mammalian host immune system by changing their major surface coat proteins, known as variant surface glycoproteins (VSG), prior to each wave of host antibodies raised against the previous VSG type. Due to this sophisticated immune evasion technique known as antigenic variation, a vaccine against the disease is unlikely in the near future. Drugs currently in clinical use are associated with severe adverse effects, difficult administration and increasing concerns regarding drug resistance. Therefore, new drugs are urgently required (Lüscher et al. Reference Lüscher, de Koning and Mäser2007). The drugs indicated for treatment of the disease (Fig. 1) depend upon the subspecies of parasite and stage of the disease.
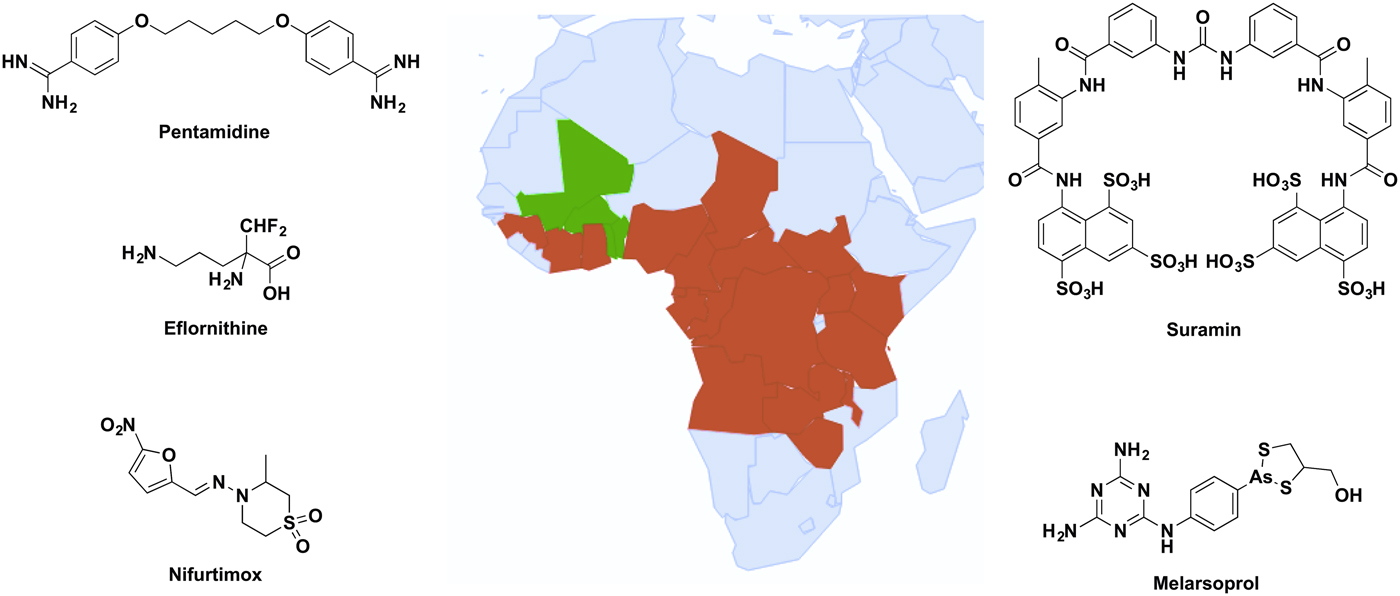
Fig. 1. Distribution of sub-Saharan countries reporting cases of T. b. gambiense and T. b. rhodesiense since 2010 according to WHO Global Health Observatory data (World Health Organization, 2016), and the clinical drugs currently used to treat HAT. Red represents cases reported, green shows no cases reported and grey indicates no data available.
Early stage T. b. gambiense is treated with pentamidine, a diamidine hypothesized to act as a trypanocidal agent through several mechanisms, including disruption of the nucleus, kinetoplast and mitochondrial membrane potential (Baker et al. Reference Baker, de Koning, Mäser and Horn2013). Late stage T. b. gambiense is treated with a combinational therapy of nifurtimox and eflornithine. Eflornithine is the only drug for HAT with a defined target, the ornithine decarboxylase, but the drug has poor potency against T. brucei and combination therapy is required to prevent drug resistance acquired by loss of the drug uptake transporter (Barrett and Croft, Reference Barrett and Croft2012). Suramin is recommended only for early stage T. b. rhodesiense due to its inability to penetrate the blood–brain barrier. Although the mechanism of uptake by the parasites is known, the trypanocidal mode of action still remains to be determined (Barrett and Croft, Reference Barrett and Croft2012; Zoltner et al. Reference Zoltner, Horn, de Koning and Field2016). The arsenical-based drug melarsoprol is recommended for late stage T. b. rhodesiense due to its ability to cross the blood–brain barrier; however, this property creates the often fatal adverse effect of encephalopathy in up to 10% of patients treated with the drug (Kuepfer et al. Reference Kuepfer, Schmid, Allan, Edielu, Haary, Kakembo, Kibona, Blum and Burri2012).
Differences in the biochemical processes between mammalian and trypanosomatid mitochondria make the mitochondrion an attractive drug target. One main difference between T. brucei and mammalian mitochondrial respiration is the presence of the trypanosome alternative oxidase (TAO), an essential non-cytochrome terminal oxidase, which has been extensively characterized as a drug target. This review will summarize the structure and function of TAO, and discuss the current progress towards the development of inhibitors against this protein.
STRUCTURE AND FUNCTION OF THE TAO
Function
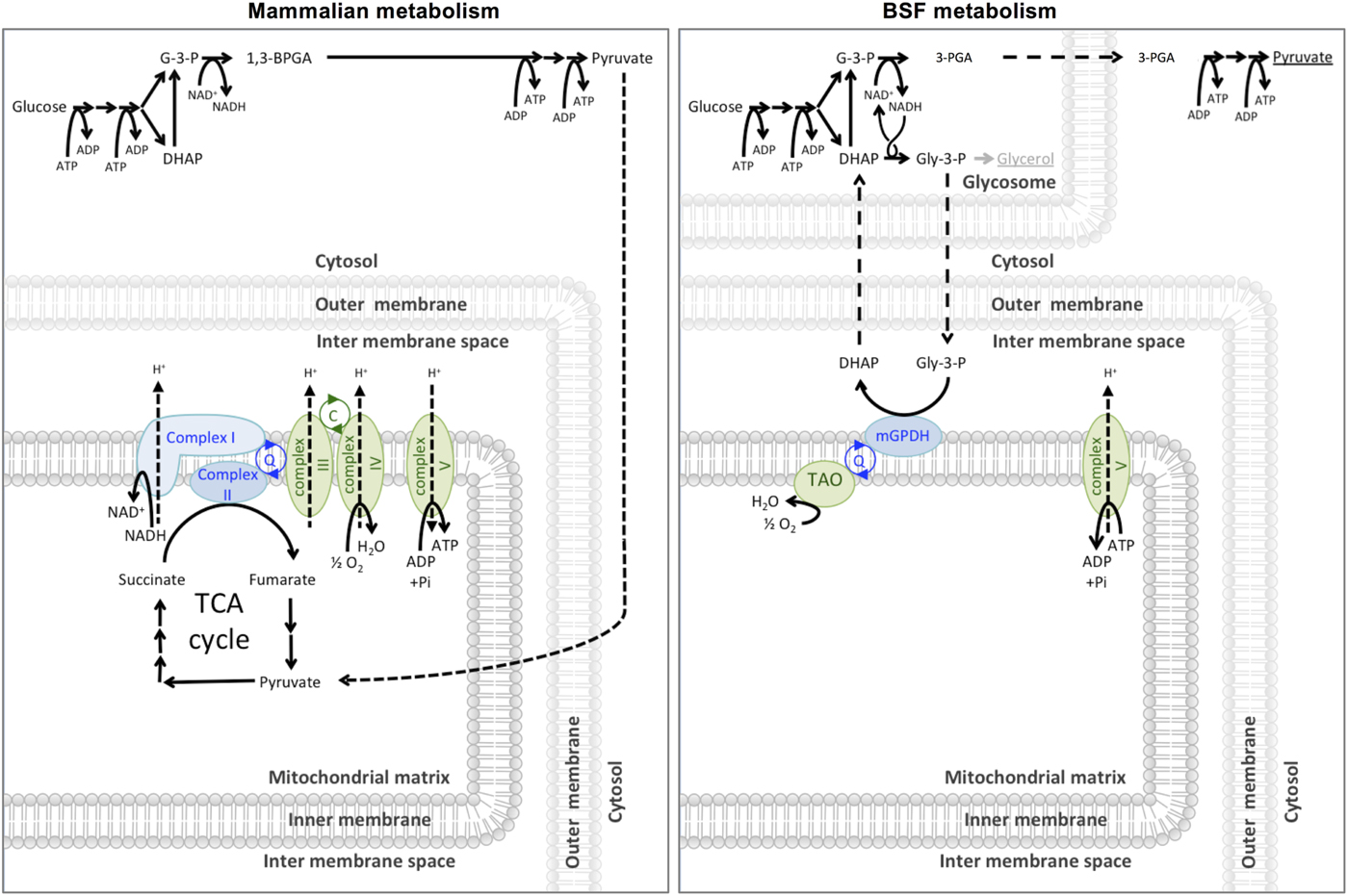
In 1960, Grant and Sargent first described the glycerol-3-phosphate oxidase (GPO) system as a cyanide-insensitive, oxygen-dependent mechanism of respiration in T. brucei rhodesiense (Grant and Sargent, Reference Grant and Sargent1960). The GPO system consists of two enzymes; a mitochondrial FAD+-dependent glycerol-3-phosphate dehydrogenase (mG3PDH) and a terminal oxidase they termed the GPO. Clarkson et al. (Reference Clarkson, Bienensb, Pollakisz and Gradyll1989) proved that ubiquinol links the dehydrogenase and oxidase of the GPO system by acting as an electron carrier, and proposed that GPO was similar to the plant alternative oxidase (AOX) and therefore should be renamed the TAO. The GPO system is responsible for the cyanide-insensitive oxygen-dependent respiration in bloodstream form T. brucei, where the GPO shuttle facilitates the reoxidation of NADH to NAD+ required for glycolysis. As shown in Fig. 2, the mG3PDH oxidizes glycerol-3-phosphate (Gly-3-P) to dihydroxyacetone phosphate (DHAP), during which four electrons are transferred to ubiquinol (Bringaud et al. Reference Bringaud, Barrett and Zilberstein2012). The electrons from ubiquinol are subsequently oxidized by TAO to convert dioxygen into water. Alternative oxidases are found across a broad range of organisms, including plants, nematodes, algae, yeast and T. brucei, but, curiously, are not known to be present in the other human-infective trypanosomatids such as T. cruzi or Leishmania spp.
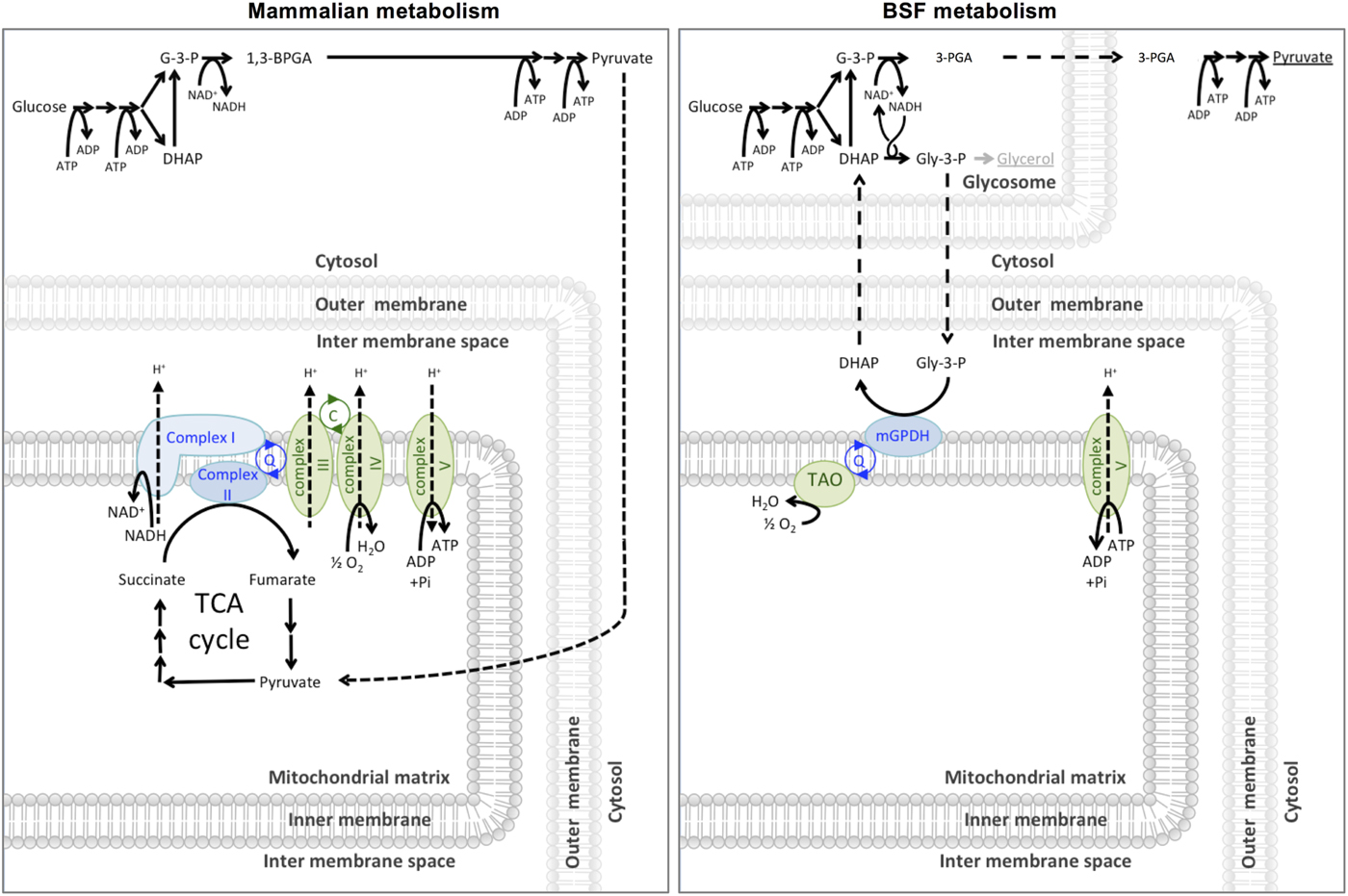
Fig. 2. Carbon source metabolism. Mammalian cells glycolytically metabolize glucose to pyruvate in the cytosol. Pyruvate is taken into the mitochondrial matrix where it is further completely metabolized to CO2 and water via the pyruvate dehydrogenase complex and the TCA (tricarboxylic acid) cycle and electron transport chain (shown in green). Entry points to the electron transport chain are shown in blue. The malate-aspartate shuttle (not shown) maintains the cytosolic NAD(H) redox. Bloodstream form (BSF) T. brucei metabolize glucose to 3-PGA in the glycosome and 3-PGA to pyruvate in the cytosol. A high rate of glycolysis means that sufficient ATP is produced through this route alone and the parasite can secrete the pyruvate produced as waste rather than spend energy consuming it further. The GPO system (mGPDH and TAO) is required to maintain glycolytic NAD(H) redox. If the GPO system is inhibited BSF T. brucei will convert Gly-3-P to the secreted end product glycerol to maintain glycolytic NAD(H) redox (Bringaud et al. Reference Bringaud, Barrett and Zilberstein2012).
TAO was first identified in T. brucei by Chaudhuri et al. (Reference Chaudhuri, Wilfred, Temple and Hill1995) using antibodies against the AOX from Sauromatum guttatum, which detected a 33 kDa protein in the parasite's mitochondria. This 33 kDa protein was subsequently purified from bloodstream form T. brucei mitochondria and confirmed to have ubiquinol oxidase activity. Chaudhuri et al. (Reference Chaudhuri, Wilfred and Hill1998) found that bloodstream form T. brucei express TAO 100-fold more than procyclic form, which is believed to be due to the ability of procyclic forms to express Complexes III and IV for ATP production via oxidative phosphorylation. Using areas of high conservation in plant AOXs, primers were designed to amplify TAO from T. brucei gDNA (Chaudhuri and Hill, Reference Chaudhuri and Hill1996). This enabled the identification of the single copy TAO gene (Tb927.10.7090) and subsequent cloning of TAO for recombinant expression in Escherichia coli (Chaudhuri and Hill, Reference Chaudhuri and Hill1996). Recombinant TAO (rTAO) was subsequently used to determine the functional activity, kinetics and inhibitors of the enzyme. Due to the endogenous ubiquinol oxidase activity of E. coli by the cytochrome bo and bd complexes, it was necessary to perform these investigations using hemA mutant E. coli, which are unable to synthesize the haeme necessary for cytochrome assembly. The ability of rTAO to restore respiration in these cells showed the ability of TAO to function as a cyanide-insensitive terminal oxidase. Research of rTAO by the Kita group established protocols for the overproduction, solubilization and purification of rTAO for use in kinetic, structural and inhibitor studies (Fukai et al. Reference Fukai, Amino, Hirawake, Yabu, Ohta, Minagawa, Sakajo, Yoshimoto, Nagai, Takamiya, Kojima and Kita1999, Reference Fukai, Coichi, Keisuke, Yoshisada and Suzuki2003; Nihei et al. Reference Nihei, Fukai, Kawai, Osanai, Yabu, Suzuki, Ohta, Minagawa, Nagai and Kita2003; Yabu et al. Reference Yabu, Yoshida, Suzuki, Nihei, Kawai, Minagawa, Hosokawa, Nagai, Kita and Ohta2003).
TAO has been implicated in several other cellular activities, such as protection against reactive oxygen species and regulation of surface protein expression. A role of AOX in photosynthetic plants is the rapid turnover of NADPH to protect the photosynthetic machinery from radicals. It is possible that TAO has a related function in T. brucei, to protect the rapidly metabolizing cells from damaging radicals. The inhibition of TAO has been shown to induce oxidative damage to proteins and increase production of reactive oxygen species (Fang and Beattie, Reference Fang and Beattie2003). Similarly, inhibition of the electron transport chain and exposure to hydrogen peroxide causes an upregulation in the expression of TAO (Fang and Beattie, Reference Fang and Beattie2003). This protection against oxidative damage may explain the ability of TAO to inhibit drug-induced programmed cell death-like phenomena in T. brucei (Tsuda et al. Reference Tsuda, Witola, Konnai, Ohashi and Onuma2006). Vassella et al. (Reference Vassella, Probst, Schneider, Studer, Renggli and Roditi2004) reported the effects of TAO inhibition on the expression levels of the procyclin GPEET, a cell surface protein found in procyclic form T. brucei. In the presence of TAO inhibitor salicylhydroxamic acid (SHAM), GPEET levels were heavily reduced leading the authors to hypothesize that the level of GPEET expression may be linked to the activity levels of TAO. Later studies showed that the expression of TAO influences the expression of GPEET, where a downregulation of both proteins may be important in the adaptation of the parasite to survive within the tsetse fly midgut (Walker et al. Reference Walker, Saha, Hill and Chaudhuri2005).
Structure
Several structures of the AOX were proposed (Andersson and Nordlund, Reference Andersson and Nordlund1999; Berthold et al. Reference Berthold, Andersson and Nordlund2000) prior to the publication of the crystal structure. Initially, hydropathy plots suggested the AOXs contain two conserved transmembrane regions, however later studies by Andersson and Nordlund (Reference Andersson and Nordlund1999) suggested AOXs are not transmembrane proteins, but rather interfacial inner membrane proteins. This was confirmed with the solving of the crystal structure of TAO (Shiba et al. Reference Shiba, Kido, Sakamoto, Ken, Tsuge and Tatsumi2013) which is devoid of any transmembrane domains, and instead has a hydrophobic face to partially bury the protein into the membrane (Fig. 3). The recent publication of the crystal structure (Shiba et al. Reference Shiba, Kido, Sakamoto, Ken, Tsuge and Tatsumi2013) should help in the design of improved TAO inhibitors. Sequence analysis of T. b. brucei, T. b. gambiense and T. b. rhodesiense showed that the amino acid sequence of TAO is identical in all three species, and therefore studies on TAO inhibitors and its co-structures can be directly applied from the common laboratory model T. b. brucei to the human disease-causing subspecies (Nakamura et al. Reference Nakamura, Fujioka, Fukumoto, Inoue, Sakamoto, Hirata, Kido, Yabu, Suzuki, Watanabe, Saimoto, Akiyama and Kita2010).

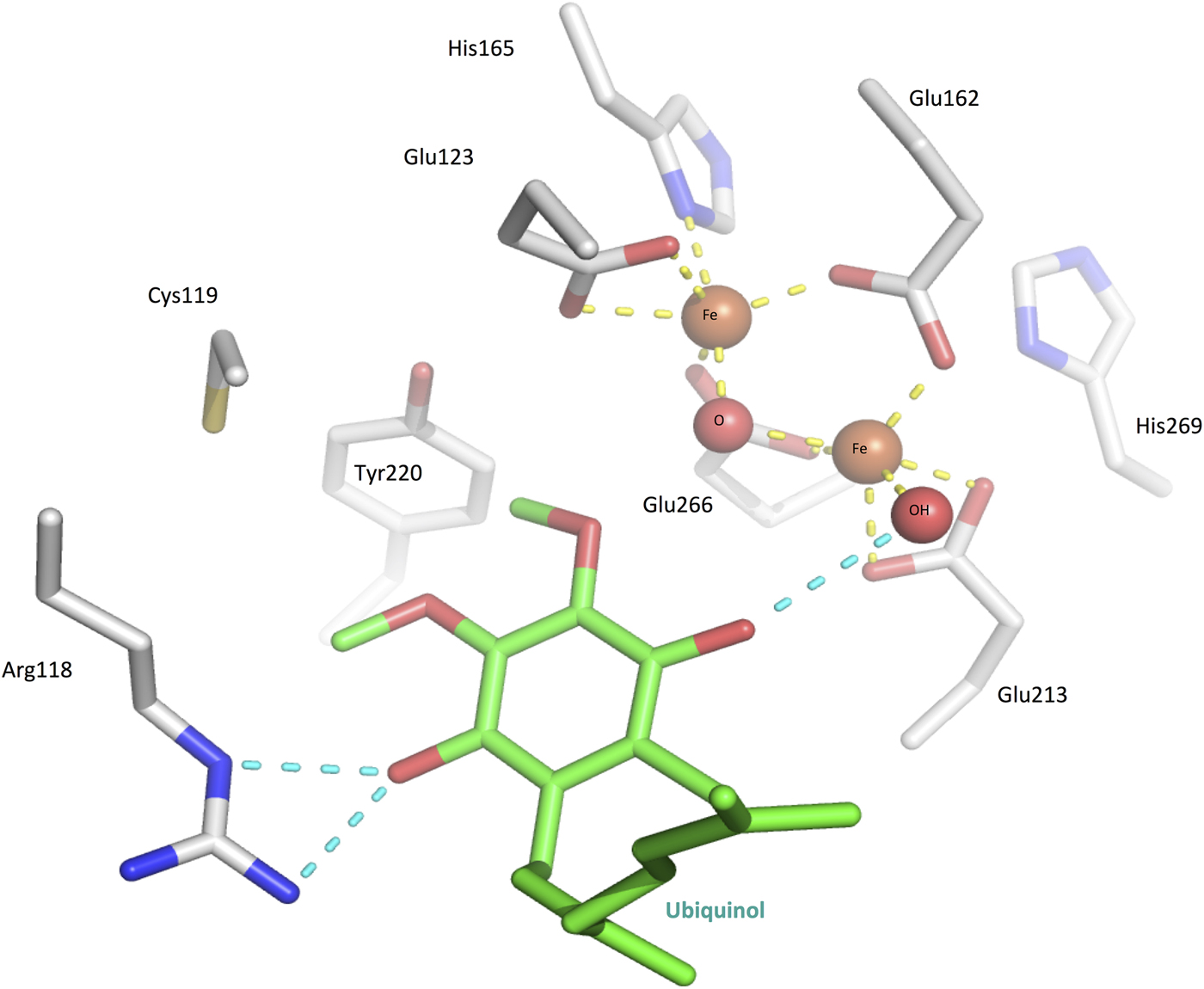
Fig. 3. The TAO dimer in complex with diiron/hydroxo core (shown as spheres) and inhibitor AF2779OH (shown in yellow carbon stick). For surface representation, hydrophobic areas are grey and hydrophilic areas are blue/red. The upper face of the dimer is highly hydrophobic allowing TAO to bury itself within a single layer of the inner mitochondrial membrane. The approximate position of the membrane/matrix interface is represented by a dashed line. The diiron catalytic core is buried deep within the protein structure, and a channel from the membrane to the core allows access of ubiquinol substrate (or analogous inhibitor AF2779OH).
Early studies on plant AOXs revealed that they were inhibited by metal chelators (Schonbaum et al. Reference Schonbaum, Bonner, Storey and Bahr1971), and subsequent investigations using EPR (electron paramagnetic resonance) (Berthold et al. Reference Berthold, Voevodskaya, Stenmark, Gräslund and Nordlund2002; Moore et al. Reference Moore, Carré, Affourtit, Albury, Crichton, Kita and Heathcote2008) and ICP-MS (inductively coupled plasma-mass spectrometry) (Kido et al. Reference Kido, Shiba, Inaoka, Sakamoto, Nara, Aoki, Honma, Tanaka, Inoue, Matsuoka, Moore, Harada and Kita2010) showed that the AOXs contained a non-haeme diiron catalytic core that was essential for catalytic activity and released during enzyme inactivation. In the crystal structure in its oxidized state, the two Fe (III) ions are coordinated in a distorted square pyramidal geometry to four glutamate residues and a hydroxo-bridge (Fig. 4). Two conserved histidines located nearby are also likely to be involved in Fe-coordination in the reduced state, as determined through Fourier transform infrared spectroscopy (FTIR) investigations (Marechal et al. Reference Marechal, Kido, Kita, Moore and Rich2009). Together, the two histidines and two glutamates form part of the two ExxH iron-binding motifs, which are common to all AOX proteins and are required for activity (Chaudhuri et al. Reference Chaudhuri, Wilfred and Hill1998; Ajayi et al. Reference Ajayi, Chaudhuri and Hill2002).
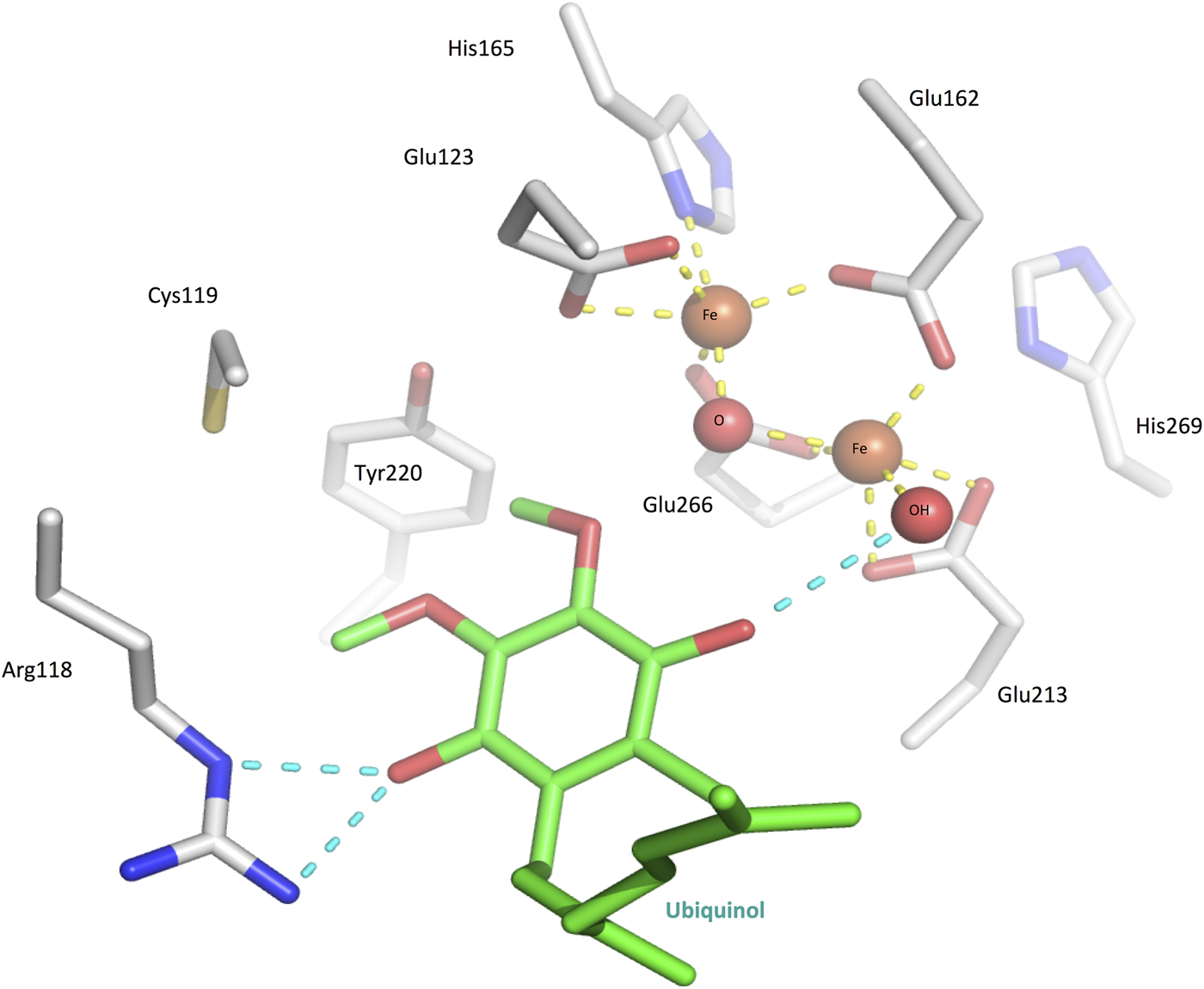
Fig. 4. The TAO active site with ubiquinol (green) superposed in the place of inhibitor AF2779OH. The diiron core is in the Fe(III/III) oxodiiron state (see mechanism part in the main body text), coordinated by four glutamates, two histidines, an oxygen and a hydroxyl (yellow dashed lines).
Mechanism of catalysis
The structure of TAO in complex with ubiquinol has not yet been solved so hypotheses regarding ubiquinol-binding have been made based upon the structure of TAO complexed with ascofuranone analogue AF2779OH. Superposition of ubiquinol over AF2779OH indicates that during catalysis a ubiquinol molecule is highly likely to occupy the same position. The molecules gain entry to the diiron active site through a relatively short (~10 Å) hydrophobic channel from the membrane-bound side of TAO (Fig. 4). In this position, the aromatic head of ubiquinol is <4·4 Å from the diiron core and is capable of forming hydrogen bonds with Arg118, Cys119 and Tyr220, all of which may be involved directly in catalysis rather than purely substrate binding.
A mechanism of catalytic activity has been proposed (Moore et al. Reference Moore, Shiba, Young, Harada, Kita and Ito2013 and Fig. 5), which begins with the diiron core in a reduced state [i.e. as Fe(II/II) bridged by a hydroxide]. Upon binding of molecular oxygen to the Fe(II/II) diiron core (Fig. 5A), one iron passes an electron to an oxygen atom, forming a superoxo intermediate comprising an oxygen radical joined to an Fe(II/III) core. The oxygen radical immediately abstracts a hydrogen atom (proton plus electron) from ubiquinol, yielding a ubisemiquinone and a hydroperoxo intermediate (Fig. 5B). The unstable intermediate then undergoes a rearrangement whereby the hydroperoxo loses its proton and electron to the hydroperoxide bridge, which is then released as water (Fig. 5C). The Fe(II/III) core then gains an interaction with one of the histidines as determined in FTIR experiments (Marechal et al. Reference Marechal, Kido, Kita, Moore and Rich2009) and the second atom of the dioxygen forming a peroxodiiron. Homolytic cleavage of the O–O bond (Fig. 5D) yields an oxodiiron core, and one of the oxygens abstracts a hydrogen atom (proton plus electron) from Tyr220 generating a tyrosyl radical, as observed by Marechal et al. (Reference Marechal, Kido, Kita, Moore and Rich2009). The tyrosyl can then pick up an electron and proton from the ubisemiquinone, either directly or via Cys119, releasing ubiquinone and returning Tyr220 to its resting state. Moore's model suggests that ubiquinol in a second channel can then provide two electrons and protons to release a second water and reduce the diiron core back to its original Fe(II/II) state bridged by a hydroxide ion through an unknown mechanism (Fig. 5E). However, the second ubiquinol channel may not be needed as the release of ubiquinone creates the space for the binding of a second ubiquinol in the same channel in a ping-pong binding fashion. Furthermore, the mechanism of electron and proton transfer could proceed through a similar route as for the first ubiquinol.

Fig. 5. Mechanism of catalysis, adapted from Moore et al. (Reference Moore, Shiba, Young, Harada, Kita and Ito2013). (A) Molecular oxygen binds to the reduced diiron core to form a superego intermediate, while ubiquinol (UQ-H2) binds to the active site. (B) The ubiquinol 4-hydroxyl is nucleophilically attacked by the oxygen radical, forming a hyrdoperoxo intermediate and ubisemiquinone. (C) The core rearranges to release a molecule of water. (D) A core oxygen picks up a hydrogen from Tyr 220 and the resulting tyrosyl radical takes the final hydrogen from the first ubiquinol, which is released as ubiquinol. (E) The core rearranges allowing a second ubiquinol to bind and react in an as-yet unknown mechanism.
INHIBITORS OF THE TAO
The effectiveness of TAO inhibition to kill T. brucei has been well debated, with conflicting historical reports in the literature as to whether the inhibition of the GPO system alone is sufficient to kill the cells. As shown in Fig. 2, bloodstream form T. brucei rely solely on glycolysis for ATP production, as opposed to the ATP-producing oxidative phosphorylation used by procyclic forms. In the presence of TAO inhibitors, the oxidation of Gly-3-P to DHAP is blocked, causing an accumulation of Gly-3-P in the glycosome, which is converted to glycerol by the ATP-producing glycerol kinase (Yabu et al. Reference Yabu, Suzuki, Nihei, Minagawa, Hosokawa, Nagai, Kita and Ohta2006). This allows the recycling of glycosomal NAD+/NADH necessary to continue glycolysis anaerobically.
Early reports of in vivo testing of TAO inhibitors suggested that although the compounds were able to inhibit the protein in vitro, this action alone was not sufficient to clear an infection when tested in animal models, due to anaerobic ATP production by the trypanosomes (Clarkson and Brohn, Reference Clarkson and Brohn1976; Grady et al. Reference Grady, Bienen, Dieck, Saric and Clarkson1993; Yabu et al. Reference Yabu, Minagawa, Kita, Nagai, Honma, Sakajo, Koide, Ohta and Yoshimoto1998). It was believed that in order to cause cell death the anaerobic production of ATP also needed to inhibited with the co-administration of glycerol. However, later investigations showed that bloodstream T. brucei exposed to TAO inhibitors alone are unable to survive for more than 24 h using only anaerobic respiration (Helfert et al. Reference Helfert, Estévez, Bakker, Michels and Clayton2001). Furthermore, subsequent studies of a TAO inhibitor with an optimized dosing regimen, but in the absence of glycerol, showed that TAO inhibition alone is sufficient to clear an infection in vivo (Yabu et al. Reference Yabu, Yoshida, Suzuki, Nihei, Kawai, Minagawa, Hosokawa, Nagai, Kita and Ohta2003), indicating that inhibition of TAO is indeed a valid drug target.
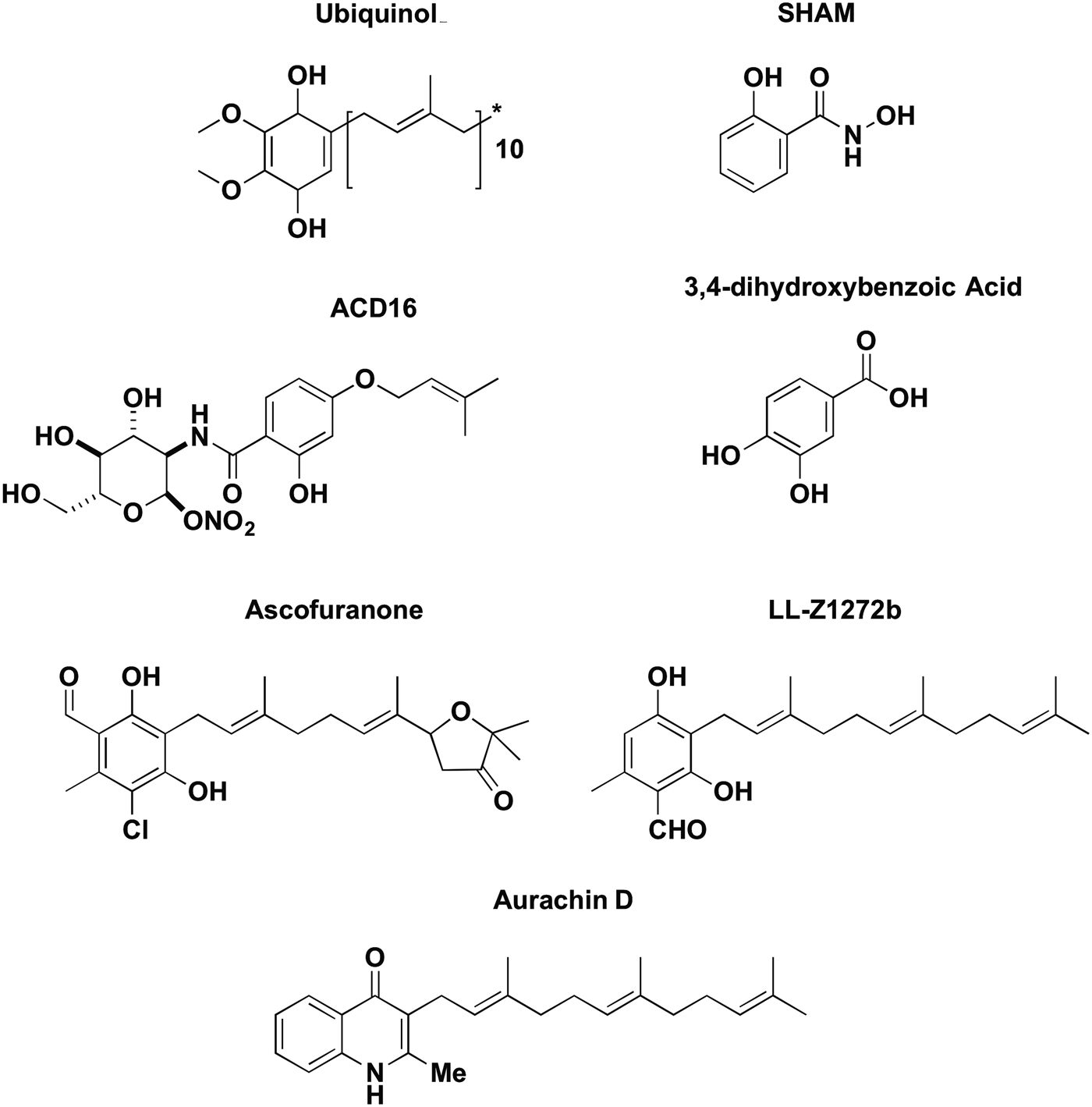
There are few compounds that have been shown to be inhibitors of TAO. These compounds (Fig. 6) all show structural similarity to the TAO substrate ubiquinol and are thought to act as competitive inhibitors, by binding to the ubiquinol binding site.

Fig. 6. Chemical structures of TAO substrate ubiquinol and the TAO inhibitors; salicylhydroxamic acid (SHAM), ACD16, 3,4-dihydroxybenzoic acid, ascofuranone, LL-Z1272 and aurachin D.
Salicylhydroxamic acid
The first compounds to be investigated as TAO inhibitors were the aromatic hydroxamates, such as SHAM (Fig. 6). The SHAM was known to be a potent inhibitor of the AOX in plants prior to the discovery of the GPO system in trypanosomes; hence, the compound was investigated as a potential inhibitor of TAO. It is thought that hydroxamic acids compete with ubiquinol for binding to TAO, and thus the compounds prevent the translocation of electrons from ubiquinol to oxygen (Pollakis et al. Reference Pollakis, Grady, Dieck and Clarkson1995). The SHAM was found to have moderate (EC50 = 15 µ m) activity against T. brucei in vitro and was shown to specifically inhibit all TAO activity at 1 mm (Opperdoes et al. Reference Opperdoes, Borst and Fonck1976), although only a little effect was seen on ATP production. However, when the trypanocidal effect of SHAM was investigated in vivo, the compound was unable to clear an infection and was only shown to be trypanocidal when co-administered with glycerol (Clarkson and Brohn, Reference Clarkson and Brohn1976).
SHAM is a poor clinical candidate, due to its low solubility in water (Nihei et al. Reference Nihei, Fukai and Kita2002), which impairs the compounds from crossing the blood–brain barrier, a critical characteristic required for drugs to effectively treat HAT. Numerous attempts were made to improve the potency of hydroxamic acids against TAO, but were unable to match the potency of SHAM when tested in vivo (Grady et al. Reference Grady, Bienen, Dieck, Saric and Clarkson1993). Recently this issue has been revisited; Ott et al. (Reference Ott, Chibale, Anderson, Chipeleme, Chaudhuri, Guerrah, Colowick and Hill2006) developed novel SHAM analogues to improve its potency and solubility. SHAM analogues such as ACD16 (Fig. 6) were designed to include a prenyl side chain, as found in the TAO substrate ubiquinol, and a carbohydrate group to improve solubility, whilst keeping the 2-hydroxybenzoic acid found in SHAM which is essential for TAO inhibition. These modifications lead to the development of three compounds with up to 5-fold greater potency than SHAM against rTAO; however, in vitro testing against T. b. brucei growth and respiration revealed none of the modified compounds were more potent than SHAM. There have been no subsequent reports on SHAM as a TAO inhibitor, although recent reports on the efficacy of TAO inhibitors without glycerol (Yabu et al. Reference Yabu, Yoshida, Suzuki, Nihei, Kawai, Minagawa, Hosokawa, Nagai, Kita and Ohta2003) may renew interest in attempts to improve upon this compound.
3,4-Dihydroxybenzoic acid
3,4-dihydroxybenzoic acids (Fig. 6) were synthesized and tested as alternative inhibitors of TAO, and displayed higher inhibitory activity than SHAM when tested in vitro, but this high potency was lost when the compounds were tested in vivo (Grady et al. Reference Grady, Bienen, Dieck, Saric and Clarkson1993). To improve the bioavailability of the compounds, a series of N-n-alkyl-3,4-dihydroxybenzamides were synthesized to increase solubility and decrease hydrolysis by serum esterases (Grady et al. Reference Grady, Bienen, Dieck, Saric and Clarkson1993). Structure activity relationships of this series of compounds showed increasing potency and decreasing solubility as the length of the alkyl substituent increases. From this, N-n-butyl-3,4-dihydroxybenzamide progressed to in vivo studies, and was found to effectively cure mice, but only when administered in conjunction with high doses of glycerol (450 mg kg−1 drug with 15 g kg−1 glycerol). Similar to SHAM, the high amount of glycerol necessary for a trypanocidal effect of N-n-butyl-3,4-dihydroxybenzamide rendered the compound unfavourable as a clinical drug candidate, and no work has been undertaken to identify if an optimized dosing regimen might clear infection in vivo without glycerol.
Ascofuranone
Ascofuranone (Fig. 6), is a biologically active natural product isolated from the fungus Ascochyta viciae. Minagawa et al. first showed that ascofuranone is a potent inhibitor of mitochondrial respiration of T. b. brucei, specifically the glucose- and Gly-3-P-dependent respiration (Minagawa et al. Reference Minagawa, Yabu, Kita, Nagai, Ohta, Meguro, Sakajo and Yoshimoto1997). Despite its high potency against TAO, ascofuranone was initially found to only be trypanocidal in the presence of glycerol, similar to the other TAO inhibitors. The minimum inhibitory concentration of ascofuranone alone was 250 µ m, whereas in the presence of 4 mm glycerol potency was improved several thousand-fold to 30 nm (Minagawa et al. Reference Minagawa, Yabu, Kita, Nagai, Ohta, Meguro, Sakajo and Yoshimoto1997). Initially in vivo testing using mouse models found that ascofuranone was only curative when co-administered with a large amount (3 g kg−1) of glycerol (Yabu et al. Reference Yabu, Minagawa, Kita, Nagai, Honma, Sakajo, Koide, Ohta and Yoshimoto1998). Despite these less than favourable initial results, the dosage of ascofuranone was improved to once again render it a promising clinical drug candidate. Yabu et al. (Reference Yabu, Yoshida, Suzuki, Nihei, Kawai, Minagawa, Hosokawa, Nagai, Kita and Ohta2003) trialled the optimal dosage to cure T. b. brucei mice without glycerol and determined that 100 mg kg−1 intraperitoneally for 4 days and 400 mg kg−1 orally for 8 days completely cleared an infection, with a 50% lethal dose (LD50) of >1·2 g kg−1 over 8 days. This study also provided evidence of the effects of ascofuranone treatment on TAO, finding that ascofuranone decreased TAO activity by 30% and increased the level of TAO expression within the cells (Yabu et al. Reference Yabu, Yoshida, Suzuki, Nihei, Kawai, Minagawa, Hosokawa, Nagai, Kita and Ohta2003).
Ascofuranone was also shown to inhibit the TAO of T. vivax, which causes animal trypanosomiasis (Nagana) in cattle. The T. vivax TAO has 76% identical amino acid residues to T. brucei TAO (Suzuki et al. Reference Suzuki, Nihei, Yabu, Hashimoto, Suzuki, Yoshida, Nagai, Hosokawa, Minagawa, Suzuki, Kita and Ohta2004) and the recombinant protein was shown to be 3-fold more sensitive to ascofuranone. Subsequent in vivo testing of ascofuranone in T. vivax infected mice found that a single intramuscular dose of 50 mg kg−1 ascofuranone without glycerol was sufficient to clear an infection, which could be reduced still further to 6 mg kg−1 over 4 days, whilst retaining 100% cure rate within 48 h. The high efficacy of ascofuranone against T. vivax may make this compound a suitable drug for use against animal trypanosomiasis.
Kinetic analysis of ascofuranone inhibition of rTAO indicated a competitive mechanism of inhibition against ubiquinol (Nihei et al. Reference Nihei, Fukai, Kawai, Osanai, Yabu, Suzuki, Ohta, Minagawa, Nagai and Kita2003). Recent studies of ascofuranone have revealed the mechanism of inhibition, interaction with TAO and the pharmacophore responsible for the inhibitory activity of ascofuranone (Saimoto et al. Reference Saimoto, Kido, Haga, Sakamoto and Kita2013). The length of the linker chain between the aromatic ring and furanone ring was shown to be important for its inhibitory activity, where the potency of inhibitor with a propyl linker was a 1000-fold lower compared with nonyl and decyl linkers. This is likely due to the interactions between the prenyl tail and membrane lipid bilayers, where hydrophobicity of the inhibitor is influenced by the length of the prenyl tail, which is important to access the membrane-associated TAO (Mogi et al. Reference Mogi, Ui, Shiomi, Omura, Miyoshi and Kita2009; Saimoto et al. Reference Saimoto, Kido, Haga, Sakamoto and Kita2013). Attempts to improve the potency and selectivity of ascofuranone-like analogues have been reported, such as the prenylphenol LL-Z1272 series by Mogi et al. (Reference Mogi, Ui, Shiomi, Omura, Miyoshi and Kita2009) (Fig. 6), although no results from in vivo testing have been reported to date.
Aurachin D
Recently the natural product aurachin D (Fig. 6), a ubiquinol oxidase inhibitor isolated from the bacterium Stigmatella aurantiaca strain Sg a15, was shown to have inhibitory activity against T. b. gambiense (Li et al. Reference Li, Herrmann, Zang, Grellier, Prado, Muller and Nay2013). Aurachin D is a mimic of ubiquinol, with a quinolone core and prenyl chain. Li et al. (Reference Li, Herrmann, Zang, Grellier, Prado, Muller and Nay2013) found that aurachin D inhibited T. b. gambiense with an IC50 of 1 µ m, with a selectivity index >35. Various analogues of aurachin D were synthesized and tested for trypanocidal activity, but none were improved compared with the natural product and hence the compound has not been taken forward into animal models.
CONCLUSIONS AND FUTURE PERSPECTIVES
Although drugs against TAO have been studied for over 40 years, there are still no drug candidates approaching clinical trials. The search for an effective TAO inhibitor has been hampered until recently by the difficulty in obtaining a crystal structure of the relatively unstable purified protein, and the historical conflicting reports on whether inhibition of TAO alone is sufficient to kill T. brucei in vivo. However, recent evidence renews the idea of TAO as a valid drug target. Although there are few inhibitors of TAO reported in the literature, it is hoped that the publication of the crystal structure of TAO will significantly improve the design of novel, potent inhibitors against the enzyme. Further work is still required to confirm the mechanism of electron transfer by TAO and that ubiquinol is the true native co-factor.
ACKNOWLEDGEMENTS
We would like to thank the rest of the members of the Gordon Florence and Terry Smith research groups.
FINANCIAL SUPPORT
This work was supported by the Leverhulme Trust (grant number RL-2012-025).